
Abstract
Background:
Iodide transport defect is an uncommon cause of dyshormonogenic congenital hypothyroidism due to homozygous or compound heterozygous pathogenic variants in the SLC5A5 gene, which encodes the sodium/iodide symporter (NIS), causing deficient iodide accumulation in thyroid follicular cells, thus impairing thyroid hormonogenesis.
Methods:
SLC5A5 gene variants were compiled from public databases and research articles exploring the molecular bases of congenital hypothyroidism. Using a dataset of 198 missense NIS variants classified as either benign or pathogenic, we developed and validated a machine learning-based NIS-specific variant classifier to predict the impact of missense NIS variants.
Results:
We generated a manually curated dataset containing 7793 unique SLC5A5 variants. As most databases compiled exome sequencing data, variant mapping revealed an increased density of variants in SLC5A5 coding exons. Based on allele frequency (AF) analysis, we established an AF threshold of 1:10,000 above which a variant should be considered benign. Most pathogenic NIS variants were located in the protein-coding region, as most patients were genetically diagnosed by using a candidate gene strategy limited to this region. Significantly, we evidenced that 94.5% of missense NIS variants were classified as of uncertain significance. Therefore, we developed an NIS-specific variant classifier to improve the prediction of pathogenicity of missense variants. Our classifier predicted the clinical outcome of missense variants with high accuracy (90%), outperforming state-of-the-art pathogenicity predictors, such as REVEL, PolyPhen-2, and SIFT. Based on the excellent performance of our classifier, we predicted the mutational landscape of NIS. The analysis of the mutational landscape revealed that most missense variants located in transmembrane segments are frequently pathogenic. Moreover, we predicted that ∼28% of all single-nucleotide variants that could cause missense NIS variants are pathogenic, thus putatively leading to congenital hypothyroidism if present in homozygous or compound heterozygous state.
Conclusions:
We reported the first NIS-specific variant classifier aiming at improving the interpretation of missense NIS variants in clinical practice. Deciphering the mutational landscape for every protein involved in thyroid hormonogenesis is a relevant task for a deep understanding of the molecular mechanisms causing dyshormonogenic congenital hypothyroidism.
Introduction
The sodium/iodide symporter (NIS) is an integral basolateral plasma membrane glycoprotein that mediates iodide accumulation in the thyroid follicular cell (1). The basolateral expression of the transporter relies on a conserved carboxy-terminal monoleucine-based sorting motif (2). The NIS-mediated iodide transport is electrogenic (2 sodium:1 iodide stoichiometry) and remarkably efficient considering the submicromolar extracellular iodide concentration (3,4).
Loss-of-function variants in the NIS-coding SLC5A5 gene cause congenital iodide transport defect, an uncommon autosomal recessive disease that results from defective iodide accumulation into the thyroid follicular cell, thus leading to dyshormonogenic congenital hypothyroidism (5). Thyroid dyshormonogenesis accounts for 10–40% of the cases of primary congenital hypothyroidism due to autosomal recessive loss-of-function variants in genes involved in thyroid hormonogenesis (SLC5A5, DUOX2, DUOXA2, TPO, TG, DEHAL1, SLC26A4, and SLC26A7) (6,7). Next-generation sequencing-based studies designed to explore the genetic bases of dyshormonogenic congenital hypothyroidism in large cohorts of patients have reported genetic variants in 20% to 60% of the subjects (8). However, a recurrent limitation relies on the criteria used to evaluate the pathogenicity of variants detected, which would have contributed to an overestimation of the prevalence of disease-causing variants (9).
Shortly after the cloning of the rat NIS cDNA (10), the first missense pathogenic variant (p.T354P) was identified in Japanese patients with congenital hypothyroidism (11,12). To date, more than thirty SLC5A5 variants have been identified in patients. The detailed molecular characterization of pathogenic NIS variants has revealed a wealth of mechanistic information about the transporter (13 –17).
Given the importance of NIS in thyroid physiology, we developed a machine learning-based NIS-specific variant classifier that outperformed state-of-the-art bioinformatics tools, thus improving the prediction of pathogenicity of missense variants. Based on our classifier, we predicted the mutational landscape of NIS underscoring that ∼28% of all single-nucleotide variants (SNVs) causing missense NIS variants are pathogenic, thus leading to congenital hypothyroidism if present in homozygous or compound heterozygous state.
Methods
SLC5A5 gene variants datasets
SLC5A5 gene variants were compiled in April 2021 from public databases: NCBI's dbSNP, NHLBI-ESP's EVS, UniProt, GnomAD, ClinVar (
Feature engineering
Discrete features: Human NIS protein information—that is secondary structure, post-translational modification sites, short linear motifs (SLiMs)—was obtained from the UniProt database (Uniprot ID: Q92911) and research articles. The NIS secondary structure model was refined according to the rat NIS homology model based on the crystal structure of the Vibrio parahaemolyticus sodium/galactose symporter (15,18). Putative SLiMs were investigated by using the eukaryotic linear motif resource.
Quantitative features: (a) Conservation score: Residue conservation was analyzed by multiple alignment UniRef50 and UniRef90 clustered sequences with ClustalO software (19), calculating the conservation score using the Jensen-Shannon divergence (19). (b) Secondary structure prediction: Secondary structure prediction was assessed by using Jpred API (20). (c) Impact of amino acid substitution: The impact of amino acid substitution was quantified by using 12 substitution scoring matrices available in AAindex2 database (21): MIYT790101, MUET020102, GRAR740104, MOHR870101, RIER950101, TUDE900101, WEIL970101, DOSZ010102, DOSZ010104, LINK010101, CROG050101, and BLAJ010101.
NIS-specific variant classifier
A total of 198 validated NIS variants (71 benign and 127 pathogenic) were used as the initial dataset to develop the classifier (Supplementary Table S2). The initial dataset was randomly split to constitute an initial training dataset, and a final testing dataset containing 10 unique variants was excluded from the training dataset. A randomized over-sampling of benign variants was generated to balance the training dataset, avoiding biased predictions. The final training dataset contains 231 variants (benign to pathogenic ratio 4:5). A set of 22 features describing each variant was acquired: 17 variables scoring residue conservation, secondary structure probability, and physicochemical distance between replaced amino acids were numerical; 5 variables describing the topological localization of the residue, the localization of the residue within or neighboring SLiMs were categorical. A Random Forest predictor to underscore the pathogenicity of missense NIS variants was developed by using the scikit-learn library (22). Hyperparameter tuning was performed by using a Randomized Search and a Grid Search obtaining the best parameters fitting the model: random_state = 342, n_estimators = 300, max_depth = 50, max_leaf_nodes = 100, min_samples_split = 2, min_samples_leaf = 2, max_features = 10; the remaining parameters were kept as the default model. Eightfold stratified K-Folds cross-validation was used to generate receiver operating characteristics (ROC) curves to evaluate the performance of the classifier.
Visualization
Figures were created by using SeaBorn Python Library (23).
Results
SLC5A5 variants dataset
We generated a manually curated dataset of SLC5A5 variants containing a total of 7793 unique variants (Supplementary Table S1). Most pathogenic variants were collected from research articles. Considering the importance of the carboxy-terminal PDZ-binding motif for NIS plasma membrane expression (24), nonsense or frameshift variants were considered pathogenic. Based on available information, only 101 variants were classified according to its clinical impact (46 benign and 55 pathogenic) (Fig. 1A). Most benign variants were synonymous (∼45%), while most pathogenic variants were either missense (∼35%), nonsense (∼15%), or frameshift (∼21%) (Fig. 1A). The majority of variants in the dataset were intronic variants of an uncertain significance (∼80%) (Fig. 1A).
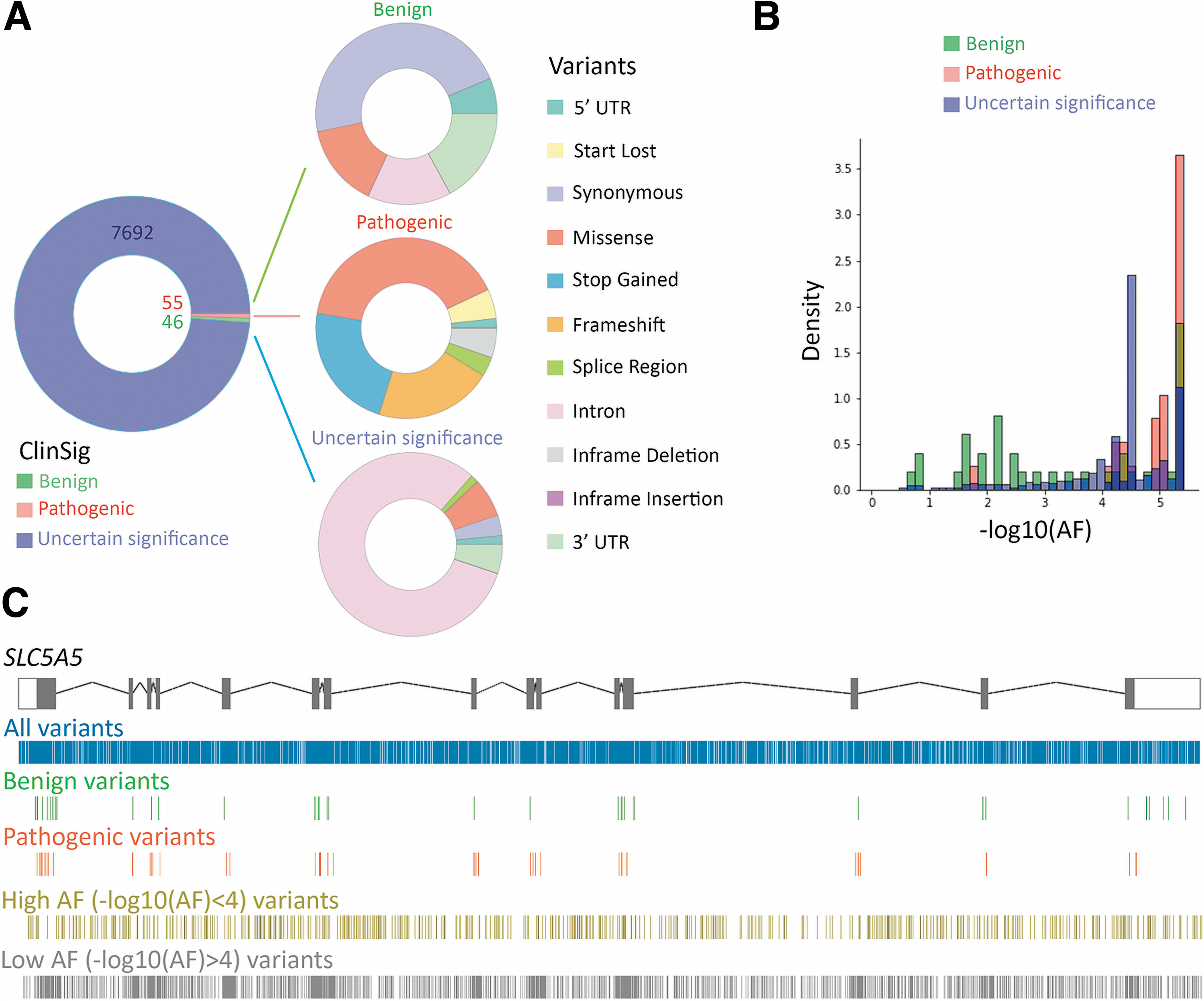
SLC5A5 gene variants. (
We analyzed the allele frequency (AF) distribution of the variants according to their clinical significance (Fig. 1B). Most pathogenic variants showed a −log10(AF) >4, thus we establish an AF threshold of −log10(AF) = 4 above which a variant might be considered benign. Using this criterion, 741 variants—mostly located in the non-coding regions—were re-classified as benign. However, the pathogenic variant c.-54C>T described in homozygous state in a Caucasian patient with dyshormonogenic hypothyroidism has an overall AF of 0.01667 in gnomAD (25).
We mapped the location of each variant along the SLC5A5 gene (Fig. 1C). Although the variants were mostly uniformly distributed along the gene, an increased density was observed in SLC5A5 coding exons (Exons 0.43 vs. Introns 0.32) as most databases compiled exome sequencing data. All pathogenic variants, except for c.-54C>T and c.970–3C>A, are located in the NIS-coding region as most patients with defective iodide transport were studied by using a candidate gene strategy limited to the sequencing of the protein-coding region. Interestingly, high AF variants were mostly mapped in non-coding regions while low AF variants were mostly represented in exonic regions.
NIS protein variants
Based on the enrichment of variants into the SLC5A5 coding region, we focused our analysis on NIS protein sequence variants. Out of the 892 variants mapped into the NIS protein sequence, only 29 variants were classified as benign, and 53 as pathogenic (Fig. 2A). The remaining set, which account for ∼90% of the total, were variants of an uncertain significance. Missense variants (∼60%) and, to a lesser extent, synonymous variants (∼33%) represent the majority of all NIS coding variants. Missense variants were classified as either benign (∼1%), of an uncertain significance (∼94%), or pathogenic (∼4%), whereas synonymous variants were either benign (∼7%) or of an uncertain significance (∼93%) (Fig. 2A). As previously observed, AF distribution of the variants revealed that pathogenic variants showed a −log10(AF) >4 (Fig. 2B).

NIS protein variants. (
The location of the variants was mapped into the secondary structure of the protein (Fig. 2C). Although the variants are mostly uniformly distributed along the protein sequence, we noticed a reduced density of variants in the transmembrane segment 3 and the cytoplasm-facing carboxy-terminus. In addition, we mapped in vitro experimentally tested NIS variants reported in the literature (Supplementary Table S2), considering benign, intermediate, or pathogenic variants as having more than 50%, 20–50%, and below 20% of wild-type NIS activity in iodide transport assays. Based on these data, we did not identify a pathogenic hotspot. However, we evidenced a tendency toward an accumulation of benign variants into regions with low sequence conservation (Fig. 2C).
A machine learning-based predictor to identify the pathogenicity of missense NIS variants
Accurate prediction of pathogenic missense variants is critical in clinical diagnosis (26). We evidenced that out of 552 missense NIS variants, 522 (94.5%) were variants of an uncertain significance. Therefore, we aimed at developing a classifier to improve the prediction of missense variants. First, we plotted benign and pathogenic NIS variants on the bases of the conservation score by using the UniRef50 and UniRef90 clusters, and the physicochemical distance between replaced amino acids according to the scoring table MIYT790101 (27) (Fig. 3A). Pathogenic variants frequently occur in highly conserved residues involving medium to high changes in the physicochemical properties of amino acids (Fig. 3A—left panel). However, benign variants appear in two clusters: one of them occurring in poorly conserved residues (low UniRef50 values), and the other one overlapping with pathogenic variants (Fig. 3A—left panel). Of note, variants of an uncertain clinical significance also appear in two clusters overlapping with benign variants (Fig. 3A—right panel). Based on these 3 parameters, only 25% of the variants of an uncertain significance showing low UniRef50 score might be predicted as benign.

NIS-specific variant classifier. (
In silico prediction tools to assess the pathogenicity of missense variants were developed by using multiple protein data, while the development of models tailored on a single protein would improve prediction accuracy (28). Therefore, we performed feature engineering and implemented a Random Forest machine-learning algorithm to develop a NIS-specific variant classifier to assess the pathogenicity of missense NIS variants. We performed eightfold cross-validation to test the algorithm and evaluated its performance assessing the area under the ROC Curves (AUC). Our classifier showed a mean AUC of 0.95 ± 0.03, with excellent sensitivity (100%) and high accuracy (90%) predicting an independent final testing dataset (Fig. 3B). The most important features were the conservation score UniRef50 and the scoring matrices RIER950101 and LINK010101 (Fig. 3C). Further, we compared the performance of our classifier with commonly used predictors such as the ensemble method REVEL, PolyPhen-2, and SIFT (29 –31). Overall, REVEL (AUC 0.91) and PolyPhen-2 (AUC 0.89) performed the best, while SIFT (AUC 0.83) was the weakest (Fig. 3D). DeLong's test for AUC comparison revealed that our classifier outperformed (p < 0.05) any single existing predictor.
Considering benign, possibly pathogenic, or pathogenic variants as having a probability below 0.5, from 0.5 to 0.7, and higher than 0.7, respectively, we investigated the clinical impact of missense variants of an uncertain significance predicting ∼68% as benign, ∼10% as possibly pathogenic, and ∼21% as pathogenic (Fig. 3E). Together, our data reinforce the importance of having a protein-specific classifier to improve the prediction of pathogenicity of missense variants.
Predicted mutational landscape of NIS
Based on our NIS-specific variant classifier, we elucidated the mutational landscape of NIS. Except M1, as start codon variants are pathogenic, the remaining 642 amino acids constituting the NIS polypeptide were individually substituted by the remaining 19 amino acids generating 12,198 missense variants. We calculated the mean probability of pathogenicity for each amino acid substitution, classifying each of them by using the aforementioned probability thresholds (Fig. 4A and Supplementary Table S3). Detailed analysis of the mutational landscape revealed that missense variants located in transmembrane segments are frequently possibly pathogenic or pathogenic, whereas missense variants located in the extracellular amino-terminal region, the first segment of the first intracellular loop, the last extracellular loop, and most of the carboxy-terminal region are mostly benign (Fig. 4A and Supplementary Table S3). The impact of variants in most intracellular or extracellular loops is variable relying on the degree of conservation of the residues and the nature of the amino acid substitution. Of note, the proximal region of the carboxy-terminus, which is highly conserved across species, revealed two pathogenic clusters associated with the disease-causing variants p.S547R and p.G561E (32,33). Although the physiological relevance of the first 10 residues of the carboxy-terminus remains to be elucidated, the variant p.G561E NIS impairs the recognition of an adjacent tryptophan-acidic motif by the adaptor kinesin light chain 2 of the motor protein kinesin-1, thus reducing NIS export from the endoplasmic reticulum (32).
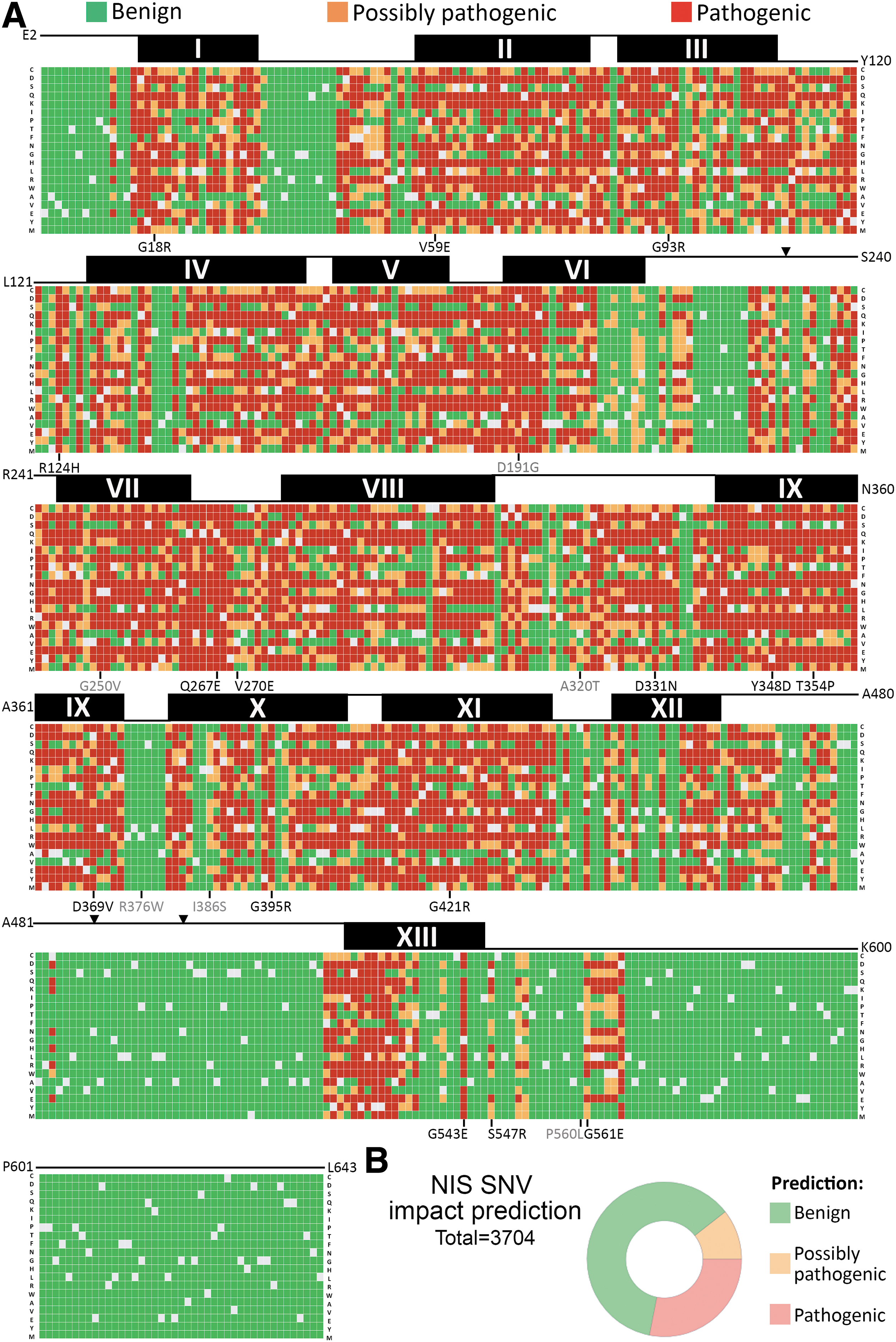
Predicted mutational landscape of NIS. (
Thereafter, we predicted the clinical outcome of SNVs, the most frequent genetic event leading to missense variants. Particularly, SNVs may cause 3704 missense NIS variants. Our classifier predicted ∼61% of these variants as benign, ∼10% possibly pathogenic, and ∼28% pathogenic (Fig. 4B and Supplementary Table S3).
Pathogenicity prediction of novel NIS missense variants
A few next-generation sequencing-based studies reported novel heterozygous missense NIS variants in patients with dyshormonogenic congenital hypothyroidism (34 –36). Considering that these missense NIS variants have not been characterized by using functional in vitro assays, we investigated their pathogenicity by using our classifier and the predictors REVEL, PolyPhen-2, and SIFT. The variants p.D191G, p.G250V, and p.I386S were consistently predicted as pathogenic, while the impact of the variants p.A320T, p.R376W, and p.P560L yielded conflicting results (Table 1).
Pathogenicity Prediction of Novel Missense Sodium/Iodide Symporter Variants
In silico predictions were carried out by using REVEL (scale: 0 = benign, 1 = pathogenic), PolyPhen-2 (scale: 0 = benign, 1 = probably damaging), and SIFT (scale: 1 = tolerated, 0 = affect protein function), and our NIS-specific classifier (scale: 0.0–0.5 = benign, 0.5–0.7 = possibly pathogenic, 0.7–1.0 = pathogenic).
NIS, sodium/iodide symporter.
The residue R376, located in the fourth intracellular loop, is mildly conserved (UniRef90 = 0.78, UniRef50 = 0.61) and predicted to participate in 2 SLiMs: a diArginine endoplasmic reticulum-retention/retrieval signal (RxxR) and a canonical 14-3-3 interaction motif (RxxSxxP). Although the variant p.R376W may disrupt any of these putative SLiMs, the effect might be minor and, thus, predicted as benign. The residue A320, located in the fourth extracellular loop, shows moderate conservation (UniRef90 = 0.79, UniRef50 = 0.71) but the variant p.A320T may not produce a significant physicochemical impact to be predicted other than benign. However, the residue P560, located in the carboxy-terminus, is highly conserved in UniRef90 cluster (0.81) but lowly conserved in UniRef50 cluster (0.58) and, thus, predicted as benign.
Discussion
We present a manually curated dataset of 7793 variants identified in the SLC5A5 gene gathered from public databases and research articles. Considering only those variants occurring in the SLC5A5 coding sequence, we compiled 893 variants, of which more than 50% are missense and ∼30% are synonymous variants. Although the analysis of synonymous variants is beyond the scope of the present work as these do not alter the corresponding amino acid residue and have no direct effect on the protein, they can affect transcription and splicing regulatory factors within protein coding regions, thus modulating gene expression and mRNA processing (37). Moreover, it is important to note that the impact of missense variants on normal pre-mRNA splicing should be considered (37). Recently, Truty et al. (38) highlighted the power of bulk RNA analysis to study in silico predicted splicing variants identified by DNA sequencing. However, the application of RNA analysis in congenital hypothyroidism might be limited, as thyroid tissue is frequently unavailable unless the patient develops suspicious thyroid nodules.
To overcome the limited information available on the clinical classification of missense NIS variants, as 522 out of 552 missense variants are of an uncertain clinical significance, we developed an NIS-specific variant classifier that allowed the prediction of the clinical outcome of missense variants with high accuracy (90%), outperforming the predictors REVEL (81%), PolyPhen-2 (81%), and SIFT (79%). The algorithm relies on the identification of pathogenic NIS variants and their functional characterization at the molecular level. We identified 198 validated missense NIS variants representing 1.62% of the mutational landscape of the protein, thus highlighting the importance of uncovering the molecular basis of a congenital disease and performing extensive structure–function analysis. Based on our classifier, we predicted that ∼28% of all SNVs putatively causing missense NIS variants are pathogenic. Significantly, several missense NIS predicted as pathogenic have a correlate in disease-causing variants identified in other sodium-coupled co-transporters members of the SLC5A family, such as SGLT-1 and SGLT-2 (39). Based on the autosomal recessive nature of the disease, we considered the benign variant as having more than 50% of wild-type NIS activity. However, the presence of mild-effect causing NIS variants should be considered in oligogenic forms of dyshormonogenic congenital hypothyroidism (40). Importantly, the contribution of NIS variants retaining residual activity to the phenotype of the patient is severely influenced by environmental conditions, such as the amount of iodide ingested in the diet (41).
Deciphering the mutational landscape of NIS is an important step forward in the interpretation of variants in clinical practice and a launching platform to further explore protein structure/function relationships. Assessing the mutational landscape of a protein of interest by using functional in vitro assays requires expensive and laborious techniques such as deep-mutational scanning or site-saturation mutagenesis (42 –45). Although the outcome of the variants is usually investigated in a model organism that facilitates high-throughput analysis, not every human protein is properly expressed in lower eukaryotes and, for those that are, the impact of a variant is not easily extrapolated to humans.
Considering that the structure of a protein is linked to its stability, function, and partner or co-factor interactions, many in silico prediction tools are based on protein structure knowledge. The impact of missense variants on protein stability has been assessed based on free-energy changes resulting from the amino acid substitution calculated from structural analysis (46,47). Unfortunately, the structure of NIS has not yet been experimentally solved, thus limiting the development or implementation of approaches relying on structural data. The analysis of pathogenic variants in globular protein structures revealed that ∼30% of them are buried from the solvent, likely leading to protein misfolding (48). Despite differences in the residue environment, the mutational landscape of NIS revealed that missense variants located in transmembrane segments are frequently pathogenic. Mechanistic studies revealed that most pathogenic NIS variants located in transmembrane segments facing the inner core of the protein are expressed at the plasma membrane but are minimally active or inactive, whereas those facing the cytoplasm or located in intracellular loops are mostly intracellularly retained (1).
A proteome-wide comparison of the distribution of missense variants revealed that, in unstructured regions, pathogenic variants frequently impact functionally relevant SLiMs residues (49). The molecular characterization of the NIS variants p.G561E and R636* revealed novel SLiMs—that is a tryptophan-acidic motif and a carboxy-terminal PDZ binding motif—whose disruption impairs NIS plasma membrane expression (24,32). Although our classifier considers the presence of SLiMs for the prediction, this feature has a minor global incidence and so may be underestimated in the calculation. Missense variants at positions 0 and −2 of the carboxy-terminal NIS PDZ domain-binding (T/S)-x-ØCOOH motif (where x at position −1 is any amino acid, and Ø at position 0 is a hydrophobic amino acid) were considered benign, whereas previous reports indicate that the identity of the amino acids at these positions serves as a determinant in the recognition of PDZ motifs (50). Further knowledge on functionally relevant SLiMs residues and tolerated substitutions will help to address the functional impact of variants on putative SLiMs.
Deciphering the mutational landscape of proteins involved in thyroid hormonogenesis is required for a deeper understanding of the molecular mechanisms leading to dyshormonogenic congenital hypothyroidism. The increasing application of next-generation sequencing technology has revealed an enormous amount of novel variants in congenital hypothyroidism-associated genes; however, the clinical impact of most variants remains unsolved. Recently, Yamaguchi et al. (34) reported novel heterozygous missense NIS variants: p.P560L, p.A320T, and p.R376W. Unlike other prediction tools, our NIS-specific classifier predicted all these variants as benign. In line, Fu et al. (51) and van Geest et al. (52) reported conflicting interpretations of missense MCT8 variants found in patients with neuropsychomotor defects without distinctive features of the Allan-Herndon-Dudley syndrome. After functional in vitro assays, several in silico predicted pathogenic MCT8 variants were downgraded to benign. Interestingly, Niroula and Vihinen (53) revealed that most widely used pathogenicity predictors have low-sensitivity predicting benign variants. Together, these facts highlight the importance of performing functional in vitro assays to underscore the pathogenicity of variants of an uncertain significance, thus allowing the development of protein-specific classifiers to be incorporated in pipelines to assess the pathogenicity of genetic variants identified in affected families undergoing high-throughput sequencing analysis.
Footnotes
Acknowledgment
The authors are grateful to Dr. Ana María Masini-Repiso (Universidad Nacional de Córdoba, Argentina) for a critical reading of the article and valuable discussions.
Authors' Contributions
M.M. and J.P.N. conceived and designed the research; M.M. acquired, analyzed, and interpreted data; J.P.N. supervised the research; and M.M. and J.P.N. wrote the article and approved the final article to be published.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This project was supported by the Fondo para la Investigación Científica y Tecnológica—Agencia Nacional de Promoción Científica y Tecnológica (PICT-2018-1596 and PICT-2019-1772 to J.P.N.). M.M. was supported by a PhD fellowship from the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET).
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3