
Abstract
Background:
The importance of thyroid hormones (THs) for peripheral body temperature regulation has been long recognized, as medical conditions such as hyper- and hypothyroidism lead to alterations in body temperature and energy metabolism. In the past decade, the brain actions of THs and their respective nuclear receptors, thyroid hormone receptor α1 (TRα1) and thyroid hormone receptor beta (TRβ), coordinating body temperature regulation have moved into focus. However, the exact roles of the individual TR isoforms and their precise neuroanatomical substrates remain poorly understood.
Methods:
Here we used mice expressing a mutant TRα1 (TRα1+m) as well as TRβ knockouts to study body temperature regulation using radiotelemetry in conscious and freely moving animals at different ambient temperatures, including their response to oral 3,3′,5-triiodothyronine (T3) treatment. Subsequently, we tested the effects of a dominant-negative TRα1 on body temperature after adeno-associated virus (AAV)-mediated expression in the hypothalamus, a region known to be involved in thermoregulation.
Results:
While TRβ seems to play a negligible role in body temperature regulation, TRα1+m mice had lower body temperature, which was surprisingly not entirely normalized at 30°C, where defects in facultative thermogenesis or tail heat loss are eliminated as confounding factors. Only oral T3 treatment fully normalized the body temperature profile of TRα1+m mice, suggesting that the mutant TRα1 confers an altered central temperature set point in these mice. When we tested this hypothesis more directly by expressing the dominant-negative TRα1 selectively in the hypothalamus via AAV transfection, we observed a similarly reduced body temperature at room temperature and 30°C.
Conclusion:
Our data suggest that TRα1 signaling in the hypothalamus is important for maintaining body temperature. However, further studies are needed to dissect the precise neuroanatomical substrates and the downstream pathways mediating this effect.
Introduction
It is long known that thyroid hormones (THs) play a crucial role in body temperature regulation and homeostasis. 1 Pathological thyroid conditions that lead, for example, to excess TH availability in the periphery result in faster metabolism and elevated body temperature. The opposite is observed in hypothyroid conditions that result in slower metabolic rates and reduced body temperature. 2
While the effects of hyper- and hypothyroidism have been classically attributed to TH action in the periphery, the importance of central actions of TH on body temperature regulation has gained attention over the past decade. Lopez et al. first described the involvement of TH in hypothalamic circuits to alter energy metabolism in the periphery. 3 However, the individual contributions of central and peripheral actions of TH for body temperature regulation are complex and not entirely understood. 1,4
The actions of TH are promoted by the conversion of circulating thyroxine (T4) to the physiological active 3,3′,5-triiodothyronine (T3) by outer ring deiodination mediated by deiodinase type 1 or 2. Subsequently, T3 acts on nuclear TH receptor α1 and β to regulate target gene expression. Both receptors are differentially expressed throughout the body, with high expression of TRα1 in the brain and muscle, while TRβ is expressed in the liver, adipose tissue, and distinct areas of the hypothalamus. However, body temperature regulation is believed to be mainly regulated via thyroid hormone receptor α1 (TRα1) action as mice heterozygous for a mutant TRα1R384C, which confers receptor-mediated hypothyroidism due to a 10-fold reduced affinity for T3, 5 display reduced body temperature at 22°C. 6 More recent studies postulated an additional contribution of central TH signaling to whole-body temperature regulation by altering the central set point, similar to pyrexia. 7,8
However, another study surprisingly did not detect body temperature changes in mice with impaired neuronal TRα1 signaling, thus questioning the role of brain TRα1 for maintaining body temperature. 9
Using mice heterozygous for the TRα1R384C mutation (TRα1+m mice), we here show that these animals exhibit a lower body temperature, which was not entirely corrected when the mice were reared at 30°C—a condition that should compensate any defect in facultative thermogenesis or tail heat loss observed previously in these animals. 6,10 Only reactivation of the mutant TRα1 by oral T3 treatment fully restored the body temperature profile in these animals. Finally, expression of the mutant TRα1R384C selectively in the hypothalamus using adeno-associated virus (AAV)-mediated delivery was found to be sufficient to induce lower body temperature at room temperature and 30°C, demonstrating that hypothalamic TRα1 signaling plays an important role for properly maintaining body temperature.
Materials and Methods
Animals
Male TRα1+m mice, 5 TRβ knockouts, 11,12 and littermate controls from those strains on a C57Bl/6NCr background were obtained from Gemeinsame Tierhaltung, University of Luebeck, Germany. Wild-type C57Bl/6NCr animals for AAV injections were purchased from Charles River Laboratories (Charles River, Germany). All mice were 3–6 months old, single-housed at 22°C on a 12-hour light/dark cycle and fed ad libitum with chow (#1314; Altromin, Germany) and water. Animals were treated with 0.5 mg/L T3 (3,3′,5-triiodo-L-thyronine; T6397; Sigma-Aldrich, Germany) in 0.01% BSA and tap water for 12 days. 8,13 Experiments were performed according to the EU guidelines (210/63/EU) and approved by MEKUN Schleswig-Holstein (Germany).
Radiotelemetry and electrocardiogram
Body temperature of freely moving mice was continuously recorded with implanted radiotelemetry transmitters and receiver plates (G2-E-Mitter and ER-4000 Receiver; PhilipsRespironics), as described previously. 13 Animals were allowed to recover for 2 weeks, before baseline recording at 22°C, 30°C, or 10°C. Electrocardiogram (ECG) was recorded using the ECGenie System (Mouse Specifics, MA).
Adeno-associated virus
C57Bl/6NCr animals were stereotaxically injected bilaterally into the hypothalamus at the coordinates relative to bregma anterior–posterior −0.10 mm, lateral–medial ±0.20 mm, and dorsal–ventral −5.50 mm using a nanoliter 2020 injector (#300704; WPI) with 250 nL of an AAV1/2 expressing either a dominant-negative TRα1 (CMV-driven TRα1R384C-IRES-mCherry, 2.81 × 1012 GC/mL) or control green fluorescent protein (GFP) (CMV-driven GFP, 2.17 × 1012 GC/mL) (VectorBuilder GmbH, Germany).
Infrared thermography
Infrared pictures of freely moving animals were taken using T335 and T540 infrared cameras (FLIR, Sweden). Before taking the infrared pictures, Vaseline was applied between the shoulder blades to reveal the skin above the brown adipose tissue (BAT). 14
Immunofluorescence staining
Brains were fixed with 4% paraformaldehyde, and free-floating 40 μm cryostat coronal sections were blocked with 5% normal donkey serum (END9010-10; BIOZOL Diagnostics, Germany) in 0.3% Triton X-100. The sections were then incubated with the primary anti-mCherry antibody (1:1000; AB0040; OriGene, Germany) or primary anti-GFP antibody (1:1000; ab290; Abcam, UK) overnight at 4°C, followed by the Alexa Fluor 594-labeled secondary antibody (1:800, A-11058; Invitrogen, Thermo Fisher Scientific, Germany) or Alexa Fluor 488-labeled secondary antibody incubation (1:800, A-21206; Invitrogen, Thermo Fisher Scientific). Animals were excluded from analysis if no or only weak hypothalamic staining of the respective marker was found.
Quantitative polymerase chain reaction
RNA isolation from snap-frozen tissues was performed following the manufacturer's instructions using RNeasy mini kits (QIAGEN, Netherlands) that include a routine DNase digestion step to prevent genomic DNA contamination. Subsequent cDNA synthesis was conducted using the RevertAid First-Strand cDNA Synthesis Kit (K1621; Thermo Fisher Scientific). Quantitative polymerase chain reaction (qPCR) was performed using GoTaq Master Mix (M7112; Promega, Germany) and the QuantStudio system (Thermo Fisher Scientific). Housekeeping genes for qPCR analysis were selected according to NormFinder for each tissue, 15 leading to Rplp0 and Hprt as the most stable combination for interscapular BAT (iBAT) and Ppia and Hprt for soleus. qPCR data were corrected for primer efficiency and analyzed using the ΔΔCT method.
T3/T4 ELISA and free fatty acid assay
Total T3 (DNOV053; NovaTec Immundiagnostica GmbH, Germany), total T4 (EIA-1781; DRG Diagnostics, Germany), and free T4 (EIA-3775; DRG Diagnostics, Germany) determinations were performed following the manufacturer's instructions. The detection limit is 0.05 ng/mL for tT3 and 0.5 ng/mL for tT4. The intra- and interassay variance for tT3 is below 11% and for tT4 is below 4.5%. Free fatty acid (FFA) concentration in iBAT was determined according to the manufacturer's instructions (ab65341; Abcam, UK).
Indirect calorimetry
Oxygen and carbon dioxide exchange in mice was measured using indirect calorimetry (TSE PhenoMaster, TSE Systems). Mice were accustomed to the open respirometry chamber before recording food and water intake and the activity profile at 20-minute intervals. Energy expenditure (kJ/day) and respiratory quotient (RQ) (RQ = carbon dioxide produced/oxygen consumed) were calculated as described previously. 16 Data analysis was performed using Excel (2021, Version: 2308; Microsoft Corporation) and TSE PhenoMaster software V6.5.3 (TSE Systems, Germany).
Western blot
Western blot was performed as described previously 17 using anti-SERCA2 (4388; Cell Signaling Technology) or anti-OXPHOS (45-8099; Invitrogen, Thermo Fisher Scientific) with secondary antibodies (P0448 and P0447; DAKO, Denmark). Band intensities were quantified using ImageLabTM Software (Bio-Rad Laboratories, Germany).
Statistics and software
Data were analyzed using GraphPad Prism 8 (GraphPad Software), MATLAB (R2018a; MathWorks, Inc.), or Excel (2021, Version: 2308; Microsoft Corporation). Information on statistical details is given in Supplementary Table S1. Figures 1A and 2A were designed using Biorender.com.
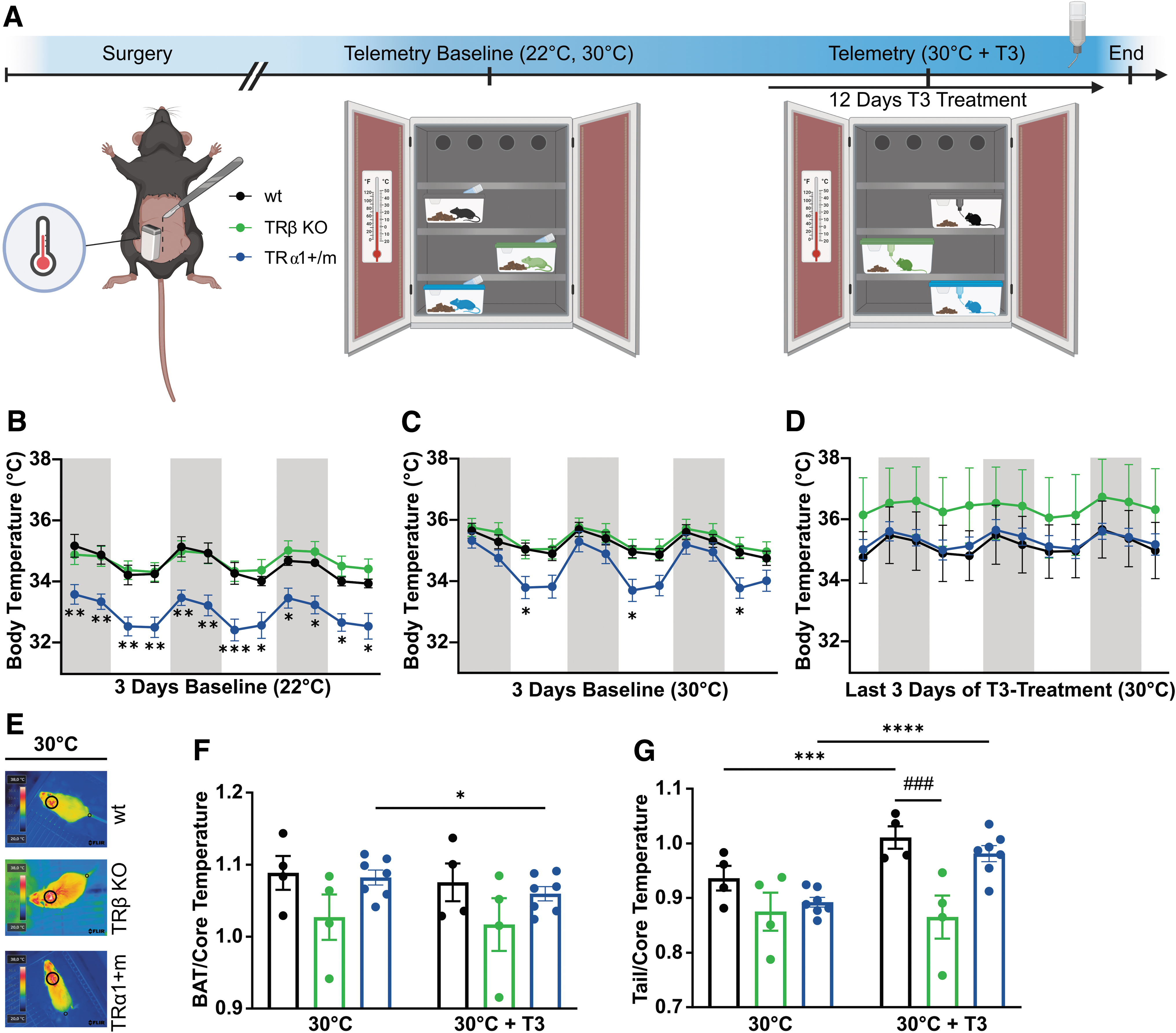
The contributions of TRα1 and TRβ to thermoregulation. (
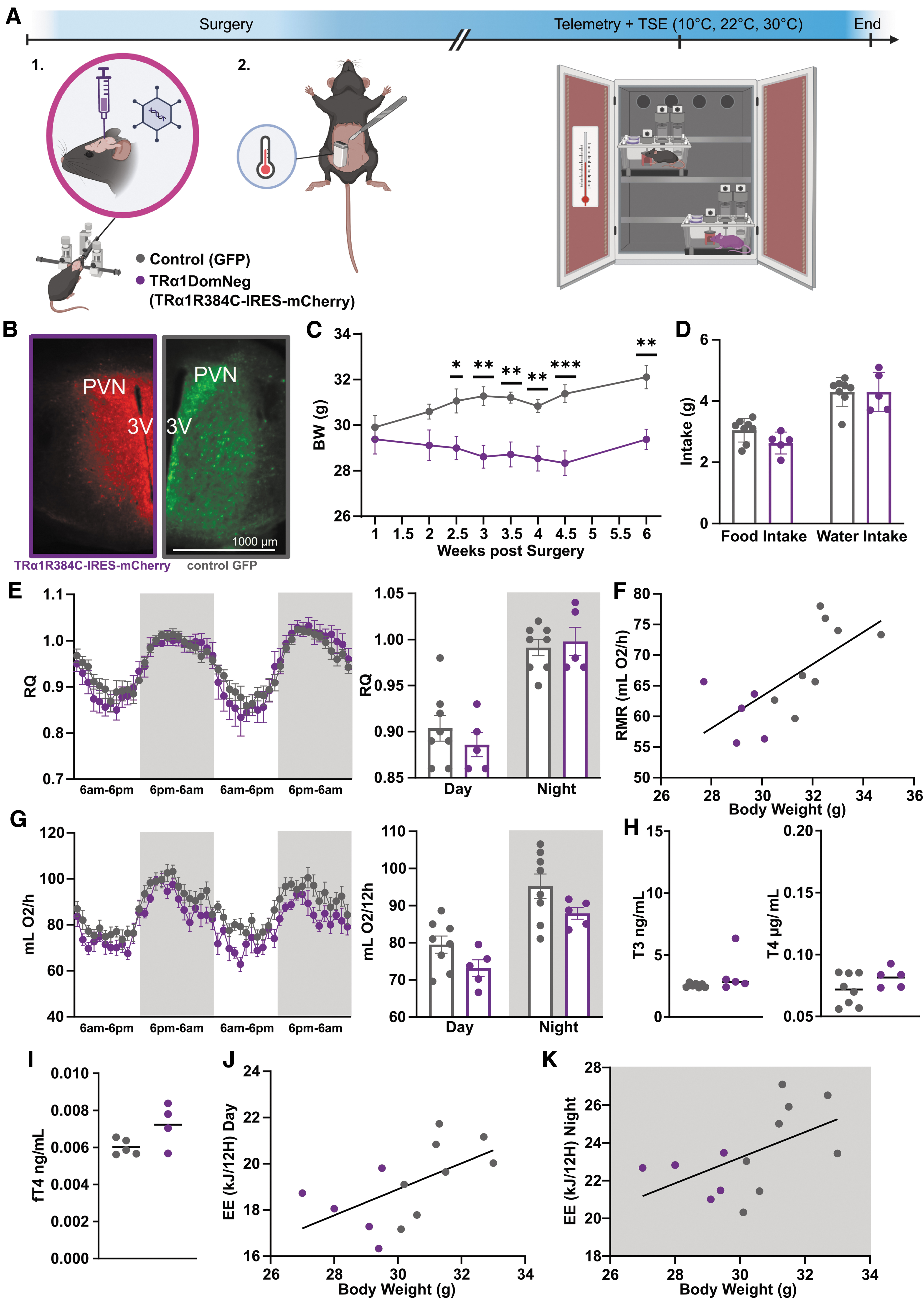
Effects of hypothalamic TRα1 on energy metabolism. (
Results
Defective body temperature phenotype of TRα1+m mice is only partially rescued at 30°C
To investigate the role of TR isoforms for body temperature homeostasis, we implanted radiotelemetry transmitters into TRα1+m mice, thyroid hormone receptor-beta (TRβ) knockout (KO) mice, and wild-type controls to record body temperature profiles. We chose a touch-free experimental paradigm, as TRα1+m mutants responded with a strong anxiety-induced elevation of body temperature to any disturbance (Supplementary Fig. S1A, B), which renders alternative methods such as rectal probes suboptimal. As TRa1+m mice have a lower body temperature at room temperature due to excessive heat loss over the tail, 6 the animals were also housed at 30°C where tail heat loss becomes negligible (Fig. 1A).
As expected, TRα1+m mice displayed a significantly 1.5°C lower body temperature at 22°C compared with controls or TRβ KO animals (Fig. 1B). When housed at 30°C to circumvent the tail heat loss of these mice, TRα1+m mutants still displayed 1.2°C lower body temperature; however, restricted to the inactive light phase (Fig. 1C). Only the reactivation of the mutated TRα1 by oral T3 treatment 5 fully normalized body temperature in TRα1+m mutants (Fig. 1D). Body temperature of TRβ KO animals was not affected at either temperature (Fig. 1B, C), and oral T3 treatment did also not lead to a significant difference compared with the wild types (Fig. 1D; Supplementary Table S2). The tail temperature of TRα1+m mice was similar to the wild types at 30°C and increased in both genotypes upon T3 treatment, whereas BAT temperature of TRα1+m mice significantly decreased by the end of T3 treatment (Fig. 1E–G).
BAT and tail temperatures were unchanged in TRβ KO animals after T3 treatment (Fig. 1E–G). On the molecular level, Adrb3 was upregulated in TRβ KO and TRα1+m mutants, while the TH-responsive genes Ucp1 and Dio2 were upregulated in TRβ KO mice but downregulated in TRα1+m mutants after T3 treatment at 30°C (Supplementary Fig. S1C). In line with other studies, 18 Dio1 and Thrsp expression was nonresponsive to T3 treatment in TRβ KO mice in the liver and kidney (Supplementary Fig. S1D, E), demonstrating the TH resistance of these tissues. Food intake increased with prolonged T3 treatment in TRa1+m mice and controls, while it remained unaltered in TRβ KO mice although on a higher baseline (Supplementary Fig. S1F, G). Water intake was comparable in all groups (Supplementary Fig. S1H, I).
Taken together, housing at 30°C and T3 treatment only led to negligible changes in TRβ KO mice, whereas without T3, TRα1+m mutants still displayed lower body temperature even at 30°C, suggesting a central resetting.
Expression of dominant-negative TRa1R384C in the hypothalamus promotes slower body weight gain
To test whether central TRα1 regulates the body temperature set point, we injected AAVs carrying dominant-negative TRa1R384C or GFP under the CMV promoter into the hypothalamus of wild-type mice, a region that is a known regulator of body temperature homeostasis, 19 combined with radiotelemetry recordings of body temperature (Fig. 2A). We observed a strong expression of mCherry (for TRa1R384C construct) or GFP (for control construct) by immunofluorescence staining in regions near the 3rd ventricle, including the paraventricular nucleus of the hypothalamus (PVN; Supplementary Table S3). Only animals with positively validated expression were included for analysis (Fig. 2B). After 1 week of recovery, food and water intake, as well as body weight and body temperature, was monitored at room temperature. Animals that received dominant-negative TRα1 stopped gaining weight and reduced their mean food intake by 0.6 g/day compared with control animals, which, however, failed to reach significance.
In comparison, control animals gradually gained weight after the surgeries (Fig. 2C, D, Supplementary Fig. S2A).
Then animals were placed into an indirect calorimetry system to assess possible changes of the RQ, energy expenditure, and activity, revealing unchanged RQ, resting metabolic rate, and metabolic profiles at 22°C (Fig. 2E–G). Since hypothalamic TH signaling is also implicated in regulating cardiovascular functions, 20 –22 we performed noninvasive ECG, which showed no differences between the groups (Supplementary Fig. S2B, C). Given that the dominant-negative TRα1 was also detected in the PVN, which regulates the hypothalamus-pituitary-thyroid axis, we tested serum total T3 and T4 levels as well as free T4 levels, which, however, remained unchanged (Fig. 2H, I). When we plotted individual energy expenditure during the active and inactive phase over the body weight of the animals, both were found to be proportional to the respective body weight at 22°C (Fig. 2J, K) as indicated by the same regression line, suggesting that the energy expenditure is weight-appropriate and not affected.
At 30°C ambient temperature, oxygen consumption gradually decreased until reaching a plateau (Supplementary Fig. S2D), while when exposed to a cold challenge (10°C) oxygen consumption gradually increased until reaching a plateau, as expected; however, with no differences between the groups (Supplementary Fig. S2D, E). Taken together, the data show minor reductions in energy metabolism with no change in locomotion (Supplementary Fig. S2F) that are, however, appropriate for the reduced body weight of the mice expressing the mutant TRα1 in the hypothalamus.
Local expression of dominant-negative TRa1R384C in the hypothalamus regulates body temperature
Most remarkably, animals that express dominant-negative TRα1 in the hypothalamus had a significantly lower body temperature at 22°C during the active and inactive phases compared with control animals (Fig. 3A). This effect persisted when placing all animals at 30°C for 6 hours (Fig. 3B) but was no longer significantly different upon cold exposure (Fig. 3B). Interestingly, normalized BAT temperature was significantly higher in dominant-negative TRα1 expressing animals, whereas tail temperature remained unchanged at 22°C (Fig. 3C, D). Nevertheless, BAT analysis did not reveal any changes in thermogenic marker expression (Fig. 3E) or FFA content (Supplementary Fig. S3A). Likewise, no browning of inguinal white adipose tissue (WAT) and epididymal WAT could be detected (Supplementary Fig. S3B, C).
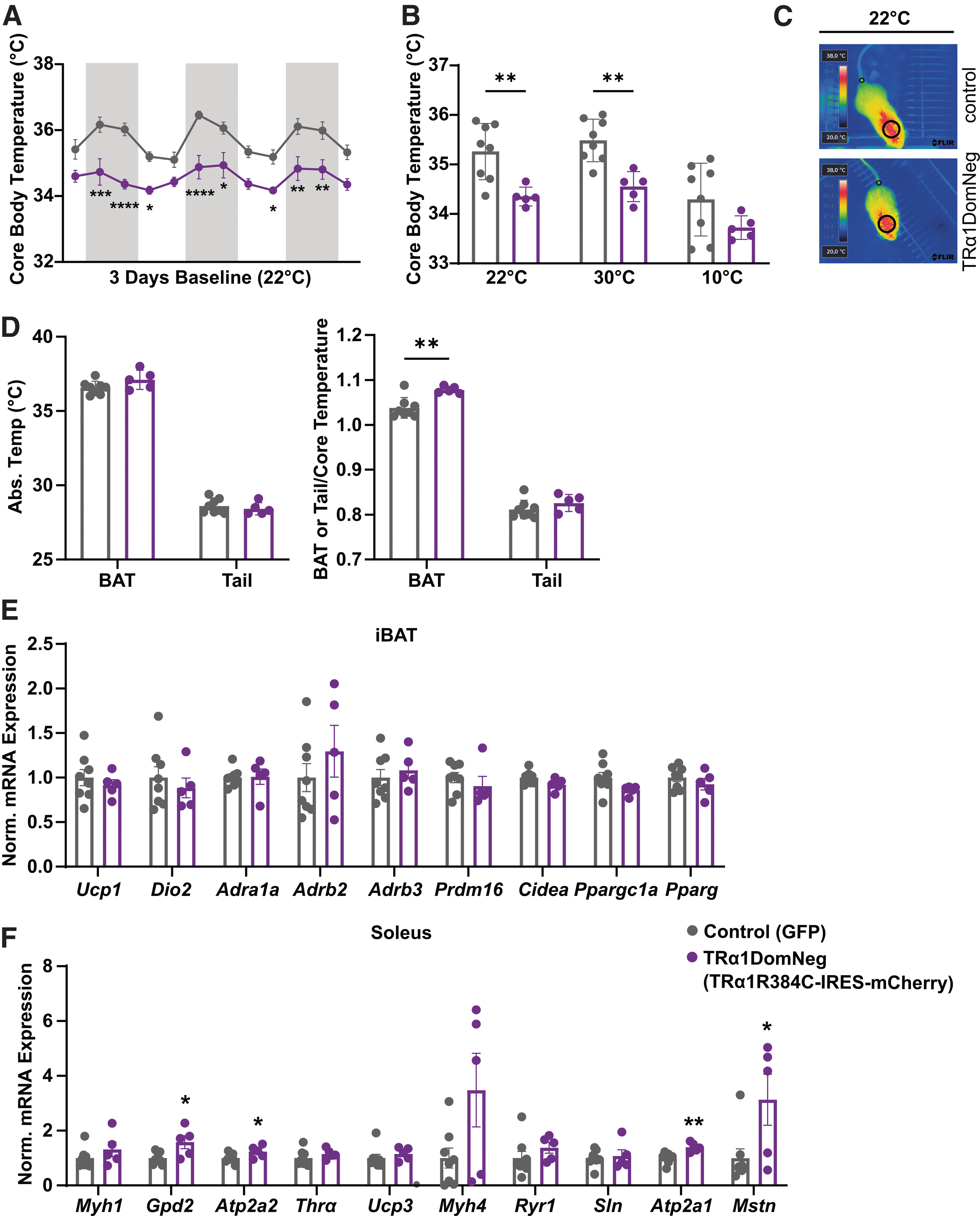
Effects of hypothalamic TRα1 on temperature regulation. (
In contrast, gene expression of soleus muscle revealed significant upregulation of Gpd2, Atp2a1, Atp2a2, and Mstn upon expression of dominant-negative TRα1 in the hypothalamus indicative of higher calcium flux and possibly impaired muscle growth (Fig. 3F), but without changes in SERCA2 or OXPHOS protein expression (Supplementary Fig. S3D, E). Gene expression in the gastrocnemius muscle remained, however, unaffected (Supplementary Fig. S3F).
Discussion
Since the individual contributions of hypothalamic TRα1 and TRβ to body temperature regulation remain enigmatic, we aimed to dissect the role of each TR in body temperature homeostasis by housing the respective mouse models at 30°C, followed by oral T3 treatment. We used radiotelemetry to track body temperature in a touch-free experimental paradigm to avoid confounding effects of anxiety-induced hyperthermia. Finally, using stereotactic delivery of AAVs carrying dominant-negative TRα1, we confirmed that the altered body temperature regulation by TRα1 originates from the hypothalamus.
Is there a role of TRβ in body temperature regulation?
The body temperature of TRβ knockout mice did not significantly differ in comparison with wild-type animals, which was unexpected given their endogenous hyperthyroidism at room temperature 11,12,23 and our previous observation that even a twofold elevated circulating T3 caused a detectable body temperature increase in wild-type animals. 21 This could be either attributed to the relatively mild hyperthyroidism in our strain with only about 60% elevation of circulating TH levels (Supplementary Table S2), or a compensatory adaptation given that the TRβ knockout is already present during embryonal development. As our study showed that body temperature in TRβ KOs responded to oral T3 treatment comparable with wild types, the first hypothesis seems more likely. Nevertheless, the tail temperature in TRβ knockout mice did not respond correctly to T3 treatment, suggesting a partial resistance to THs in heat dissipation.
Although the involvement of hypothalamic TRβ in body weight regulation and energy metabolism has been proposed previously, 24 our data therefore suggest that the role of TRβ in body temperature regulation seems to be restricted to minor defects in heat dissipation, and that the thermoregulatory phenotype of TRβ knockout mice primarily depends on their degree of hyperthyroidism.
Normalization of body temperature in TRa1+m mutants requires reactivation of TRa1
Many mouse experiments are conducted at room temperature; however, in contrast to humans, mice are constantly exposed to a mild cold challenge at room temperature that causes permanent sympathetic signaling and BAT activation. Consequently, recent studies suggest that thermoneutral housing at 30°C may be translationally more relevant. 25
As previously shown, TRα1+m mice display a significantly lower body temperature due to excessive tail heat loss as a result of defective tail vasoconstriction at room temperature. 6 By placing the TRα1+m mice at 30°C, we circumvented the excessive tail heat dissipation. Interestingly, however, the body temperature of these animals was still not entirely rescued, suggesting that additional mechanisms are at play; particularly in the resting animals, when locomotor activity did not contribute to thermogenesis. This could point to a possible defect in resting skeletal muscle, which is a main TRα1 target tissue 2,26 and involved in mediating the increase in energy expenditure upon T3 treatment. 27
It is therefore conceivable that a defect in muscle metabolism caused by the mutant TRα1 might impair muscle thermogenesis; however, cold exposure of TRα1+m mutant mice of up to 6 hours—a time period in which facultative thermogenesis is almost exclusively governed by muscle shivering—did not lead to derailed body temperature, suggesting that muscle thermogenesis is not severely affected in those animals. 10 Most interestingly, as we did not observe a compensatory response in BAT or tail temperature as a possible adaptation to maintain the body temperature, we conclude that the central circuits to maintain body temperature are altered by the mutant TRα1. Only oral T3 treatment, which led to a reactivation of the mutant TRα1, fully normalized the body temperature difference of TRα1+m mice at 30°C.
This unequivocally demonstrates that intact TRα1 signaling is required for proper body temperature regulation by excluding a permanent developmental thermoregulatory defect in these animals, which was recently observed for cardiovascular function. 13,28
Hypothalamic TRa1R384C expression suffices to reduce body temperature
A primary hypothalamic function is the regulation of energy uptake and expenditure. 29 A recent study showed that neuronal TRα1 mutants had lower energy expenditure, 9 while our mice expressing a dominant-negative TRα1 selectively in the hypothalamus displayed normal energy expenditure yet suffer from the inability to gain weight.
There are, however, a few differences between these two models that could account for the difference. First, the neuronal mutant affects the entire brain, including other regions such as the brain stem, also affecting the metabolic rate. 30 Second, the neuronal model is based on a Cre-lox approach, which triggers the expression of the mutant TRα1 already during development in the majority of neurons, while our mice express the transgene only in the adult hypothalamus, but not necessarily limited to neurons. While this allows assessing specific neuroanatomical regions, it still is far from a distinct cellular population, thus likely resulting in a mixture of effects.
This could, for instance, include actions in the ventromedial hypothalamus, where impaired TRα1 signaling is known to modulate downstream BAT action, 3 the preoptic area, which harbors thermoresponsive GABAergic neurons, 31 or the arcuate nucleus that controls energy expenditure and even myostatin expression, 32 which we also found to be altered in the soleus muscle. Consequently, further studies combining the AAV approach with a Cre-lox system need to be conducted to identify specific cellular contributions.
Most importantly, the expression of hypothalamic dominant-negative TRα1 was sufficient to reduce body temperature at room temperature and at 30°C, while the difference was not as pronounced at 10°C. The phenotype therefore shows some aspects of the expected anapyrexia from lower TH signaling, 7,8 but upon acute cold, other thermoregulatory mechanisms seem to partially mask this effect. Interestingly, this contrasts previous findings, where the neuronal expression of a mutant TRα1 did not lead to reduced body temperature at room temperature. 9 However, given that TRα1+m mice exhibit a high anxiety, 33,34 and any kind of disturbance such as simply opening the cage causes an immediate stress-induced hyperthermia, the rectal probing used to record body temperature in the neuronal TRα1 mutants 9 would not have been able to accurately record reductions in body temperature.
Conclusion
Taken together, our results demonstrate that impaired hypothalamic TRα1 signaling interferes with body temperature regulation, potentially by lowering the body temperature set point. However, further studies targeting specific hypothalamic subpopulations will be required to dissect the individual and possibly interacting contributions of the different neuroanatomical regions. Furthermore, while our data may point to the muscle rather than tail and brown fat as a thermoregulatory culprit for the lower body temperature, the precise mechanisms remain to be discovered.
Footnotes
Acknowledgments
We thank Christina Ehrengut and the staff of the GTH of the University of Lübeck for excellent animal caretaking and Julia Resch for technical assistance.
Authors' Contributions
S.C.S., R.D., R.O., and B.K. conducted the experiments; S.C.S., R.D., and R.O. analyzed the data; S.C.S., K.A.I., and J.M. designed the study; S.C.S. and J.M. drafted the first version of the article; all the authors read, corrected, and approved the final version of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants of the Deutsche Forschungsgemeinschaft (GRK1957 Adipocyte Brain Crosstalk and the TRR296 “LocoTact” Project-ID 424957847 and 445465132 to J.M., as well as Project-ID 434396546 to R.O.).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3