
Abstract
Background:
Brain–lung–thyroid syndrome (BLTS) is caused by NKX2-1 haploinsufficiency, resulting in chorea/choreoathetosis, respiratory problems, and hypothyroidism. Genes interacting with NKX2-1 mutants influence its phenotypic variability. We report a novel NKX2-1 missense variant and the modifier function of TAZ/WWTR1 in BLTS.
Methods:
A child with BLTS underwent next-generation sequencing panel testing for thyroid disorders. His family was genotyped for NKX2-1 variants and screened for germline mosaicism. Mutant NKX2-1 was generated, and transactivation assays were performed on three NKX2-1 target gene promoters. DNA binding capacity and protein–protein interaction were analyzed.
Results:
The patient had severe BLTS and carried a novel missense variant c.632A>G (p.N211S) in NKX2-1, which failed to bind to specific DNA promoters, reducing their transactivation. TAZ cotransfection did not significantly increase transcription of these genes, although the variant retained its ability to bind to TAZ.
Conclusions:
We identify a novel pathogenic NKX2-1 variant that causes severe BLTS and is inherited through germline mosaicism. The mutant lacks DNA-binding capacity, impairing transactivation and suggesting that NKX2-1 binding to DNA is essential for TAZ-mediated transcriptional rescue.
Introduction
Brain–lung–thyroid syndrome (BLTS) is a rare disorder consisting of chorea/choreoathetosis, respiratory problems, and hypothyroidism. It is caused by heterozygous mutations in the transcription factor NKX2-1 (formerly TITF1), which is involved in the development of the basal ganglia, lung, and the thyroid gland. 1,2 Human NKX2-1 defects are monoallelic and predominantly described as de novo or inherited in an autosomal dominant (AD) fashion. Rarely, some reports indicate gonadal mosaicism.
Early diagnosis of BLTS can be challenging owing to its high clinical variability and asynchrony. Pulmonary dysfunction may manifest as neonatal respiratory distress or airway infections, and severe cases are fatal. 3,4 Thyroid dysfunction may be diagnosed as congenital hypothyroidism or later as isolated hyperthyrotropinemia or complete hypothyroidism. 5,6 Neurological defects may present as hypotonia and motor retardation, with childhood chorea often being a key indicator of the disease. Athetosis and intellectual impairment may also develop during follow-up. 7
The clinical variability of BLTS has been linked to modifier genes that influence the penetrance and severity of the phenotype. 8 –10 Such genes may have tissue-specific expression, modifying specific syndrome phenotypes. In the thyroid, NKX2-1 functionally cooperates with the transcription factor PAX8 5,11,12 and other proteins (p300, DREAM, BR22, and TAZ). 13 Among these, the transcriptional coactivator TAZ, a PDZ-binding protein, plays an important role in linking transcription factors to the basal transcriptional machinery in target genes. 14,15 TAZ interacts with NKX2-1 in vitro 16 and cooperates with mutant NKX2-1 to overcome transcriptional deficiencies in a variant- and promoter-specific manner. 13
While TAZ fully rescued the in vitro transcriptional capacity of a C-terminal frameshift variant of NKX2-1 on a lung-specific promoter, it had no effect on an N-terminal frameshift variant, suggesting that TAZ modifies the respiratory phenotype in BLTS. The only requirement identified to date for TAZ to efficiently rescue the transcription of NKX2-1 mutants is that they retain some residual DNA-binding capacity. 13 However, it is currently unknown whether specific mutations in the NKX2-1 DNA-binding homeodomain are responsive to the restorative effects of TAZ.
In this study, we identify a novel pathogenic NKX2-1 homeodomain missense variant segregating within a large BLTS family and examine its phenotypic expression over three generations. We also assessed its ability to interact with DNA, transactivate three tissue-specific promoters, and whether its functional deficiency can be rescued by TAZ.
Materials and Methods
Genomic DNA mutation testing was performed by in-house-designed next-generation sequencing (NGS) using a 390 thyroid gene panel (Thyroseq-V2). 17 Segregation analysis of an NKX2-1 variant was performed in 12 family members. DNA from lymphocytes, saliva, and hair follicles of the grandparents was pyrosequenced, and paternity testing for microsatellites was performed to confirm the mode of inheritance using the Devyser Compact v3 Kit (Devyser, Stockholm, Sweden), which contains multiple chromosome-specific polymorphic repeat sequences (short tandem repeats) located on chromosomes 13, 18, 21, X, and Y, used for the analysis of the most common aneuploidies. The Declaration of Helsinki was followed, and the study was approved by the Ethics Committee of the La Paz University Hospital. Informed consent was obtained from all subjects or their legal guardians.
Human mutant NKX2-1 cDNA (p.N211S) was generated by site-directed mutagenesis and subcloned into the pcDNA3 vector. Its function, expression, and cellular localization were analyzed by promoter activity, electrophoretic mobility shift assay (EMSA), western blotting, immunofluorescence, and proximity ligation assay (PLA) (see Supplementary Data S1). The primers and antibodies used are listed in Supplementary Tables S1 and S2.
GraphPad Prism software v5 (San Diego, CA, USA) was used for statistical analysis. Student’s two-tailed t-test was used to assess the differences between measurements, with p < 0.05 considered statistically significant.
Results
Patients
The proband was born to Caucasian nonconsanguineous parents in an early-term pregnancy, with anthropometric parameters of weight 2190 g (p < 3), length 46.5 cm (p3–10), and head circumference 32 cm (p10–25). Mild hypotonia and congenital hypothyroidism were noted at birth, with filter paper thyrotropin (TSH) of 25 mU/L (normal <10 mU/L), serum TSH of 45.6 mU/L (0.50–5.40), and free thyroxine of 0.80 ng/dL (0.89–1.76). Thyroid ultrasound revealed a slightly reduced gland size. Treatment with levothyroxine (13 µg/kg/day) was started, with rapid normalization of TSH, with a current dose of 1.5 µg/kg/day at the age of 14 years. He was diagnosed with severe bronchiolitis at 3 months of age, requiring domiciliary oxygen for 6 months. He later developed chronic interstitial lung disease and was treated with inhaled bronchodilators (Fig. 1A).
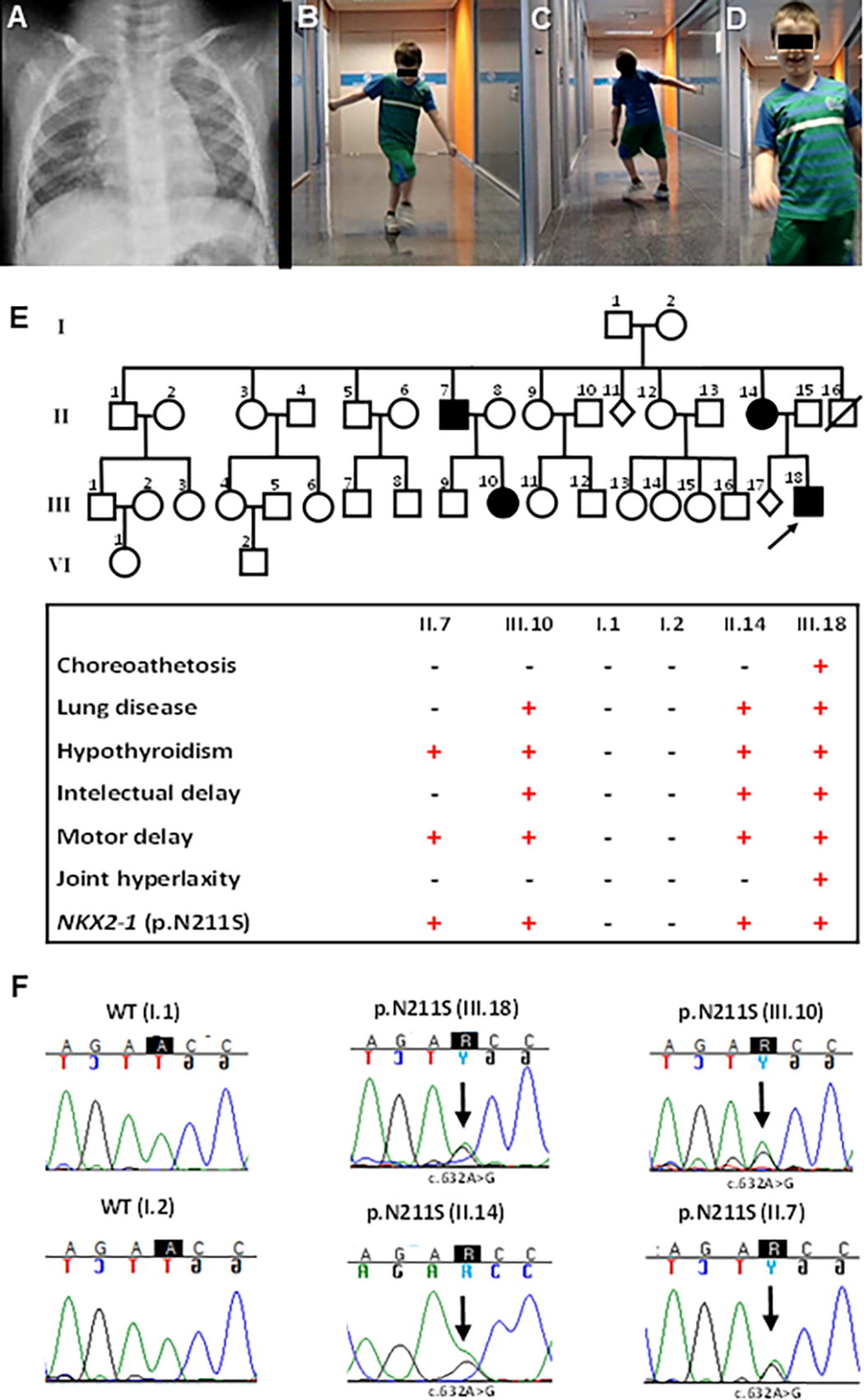
Clinical and genetic findings in a BLTS pedigree.
Developmental motor delay and poor coordination with frequent falls were also observed in early infancy. Choreic and dystonic movements were noted at 3 years of age and have been prominent since then (Fig. 1B, C). Brain magnetic resonance imaging, electromyogram, and metabolic studies were normal. His craniofacial phenotype included a broad forehead with frontal bossing, a short anteverted nose, and a poorly formed philtrum (Fig. 1D). At the age of 8, he was diagnosed with attention-deficit disorder and treated with methylphenidate with an adequate response and mild improvement of his chorea. He currently attends a mainstream school but requires speech therapy and individual support in most curriculum subjects, because of impairments in fine motor activities and writing. He also has joint hypermobility, obesity, hypercholesterolemia, and insulin resistance.
His mother had a severe respiratory infection at 7 days of age, followed by a diagnosis of asthma and motor delay in infancy. Hypothyroidism was diagnosed during pregnancy and is being treated. She currently presents with tremor, mild chorea, overweight, hypercholesterolemia, and a nonfunctioning parathyroid adenoma. His maternal uncle has a history of hyperthyrotropinemia and motor delay. His 17-year-old maternal cousin had recurrent respiratory infections in infancy and delayed psychomotor development but not chorea. She currently has asthma, hypothyroidism since the age of 9, precocious puberty, and short stature. Remarkably, the maternal grandparents were healthy, with normal motor skills, thyroid profile, and cognitive and respiratory function (Fig. 1E).
Identification of the NKX2-1 mutation
Sequencing of NKX2-1 revealed a novel missense variant c.632A>G (NM_003317.3), resulting in an amino acid change at position 211 (p.Asn211Ser; p.N211S), located in the DNA-binding domain (Fig. 1F). The pathogenic profile of the variant is characterized in Supplementary Tables S3 and S4. The genetic defect was present only in the proband (III.18), his mother (II.14), his maternal uncle (II.7), and his cousin (III.10), all with clinical variability of BLTS, particularly regarding the neurological phenotype (Fig. 1E, 1F). The proband was wild type (WT) for the TAZ and PAX8 coding sequences.
Germline mosaicism as an inheritance mechanism for the NKX2-1 variant
To investigate the possibility of mosaic inheritance of the NKX2-1 defect, DNA from saliva and hair follicles was obtained from the grandparents and analyzed by sensitive pyrosequencing, and deep-reading NGS was performed on lymphocyte DNA.
The family genotype established AD inheritance of the variant between members of the second and third generations. The microsatellite test confirmed the paternity of the grandparents, but pyrosequencing and deep NGS (>500 reads/nucleotide) showed that the p.N211S variant was absent in the three DNA samples available from the grandparents. This information supports germline (gonadal) mosaicism as the mode of inheritance from the first generation (Supplementary Fig. S1). Sperm donation was suggested to the grandfather for investigation, but the request was declined.
Functional analysis of p.N211S variant
Using oligonucleotides containing the NKX2-1 consensus DNA-binding sequence of two target genes, TG and SFTPB, we observed that the p.N211S variant failed to bind DNA (Fig. 2A, lane 6), whereas WT NKX2-1 did (Fig. 2A, lane 3). This establishes the pathogenic causal relationship between the p.N211S variant and the occurrence of BLTS in this family. This was also confirmed by transfection experiments, in which WT NKX2-1 cDNA increased the basal transcription of the target promoters TG, LHX6, and SFTPB 4- to 28-fold, whereas the p.N211S variant decreased this transcriptional activity (Fig. 2B). Cotransfection with TAZ did not increase transactivation of p.N211S NKX2-1 on any of the three promoters examined (Fig. 2B). Expression of the transfected vectors was confirmed by western blotting and their nuclear localization by immunofluorescence (Fig. 2C, 2D). Furthermore, a PLA revealed the interaction between TAZ and NKX2-1 in both WT and variant forms (Fig. 2E). The inability of the p.N211S mutant to bind DNA completely prevented any functional rescue by TAZ despite the fact that it contained intact amino-terminal and carboxyl-terminal transactivation domains (N-TAD, C-TAD), which have previously been shown to be involved in the interaction between NKX2-1 and TAZ 13 (Supplementary Fig. S2).

Functional analysis of the p.N211S mutation in NKX2-1.
Discussion
BLTS is a group of thyroid, respiratory, and neurological manifestations caused by defects in NKX2-1, with high clinical variability. 3,6 The heterogeneity of the phenotype is not fully understood. Although it appears to be more severe when the mutations are in the homeodomain, 18 this has not been confirmed in large clinical series, 19,20 suggesting that other factors underlie the phenotypic variability.
NKX2-1 variants in humans are monoallelic, likely because of the lethal consequences of embryonic disruption of the gene, as occurs in mice. 21 Therefore, NKX2-1 defects are inherited dominantly, 22 –24 or arise de novo. Transmission by germline mosaicism is rare, 25 –27 and the present study provides a compelling familial case in which the grandparents were healthy, but the NKX2-1 defect in four symptomatic offspring strongly suggests this mode of inheritance. In the literature, two mosaic individuals have been described as asymptomatic, suggesting that a late (or germline-specific) mutation occurred in the embryo before brain, lung, or thyroid differentiation. 25,26 By contrast, a third mosaic individual presented with chorea and hypothyroidism despite normal genetic analysis for the 14q deletion that caused BLTS in two offsprings. 27 Because of the typical difficulties in detecting it, the prevalence of somatic mosaicism may be underestimated in founders of BLTS families with presumed de novo NKX2-1 defects, which actually account for 30–40% of cases. 5,6,28
The main pathogenic mechanism in BLTS is haploinsufficiency; however, some NKX2-1 mutants show a dominant-negative effect in a tissue-specific manner. 29,30 This suggests the involvement of other tissue-specific proteins that interact with NKX2-1 and modulate its transcriptional capacity. Indeed, modifier genes that control the presence or severity of symptoms in BLTS have been shown to underlie clinical variability in some tissues. 13,31
TAZ is a transcriptional coactivator and a 14-3-3-binding molecule 14 whose expression is almost ubiquitous, with high levels in the lung, where it plays a role in its development. TAZ also regulates thyroid cell differentiation, participating in the Hippo signaling pathway. 32 –34
TAZ rescues the in vitro transcription of an NKX2-1 C-terminal mutant from a lung-specific promoter. 13 Contrastingly, it failed to rescue a similar NKX2-1 mutant located N-terminally with an intact N-TAD but an aberrant homeodomain sequence. Based on molecular data, we have previously described a mechanistic model for the conditions that allow transcriptional rescue of NKX2-1 defects by TAZ. 13 We hypothesized that NKX2-1 mutants must retain at least some residual DNA-binding capacity for TAZ to support transcription (Supplementary Fig. S2). As we have previously shown by coimmunoprecipitation, TAZ interacts with NKX2-1 through both N- and C-TADs; 13 therefore, the novel p.N211S mutant, lacking DNA-binding capacity but retaining intact N- and C-TADs, provided an excellent challenge to the model (Supplementary Fig. S2). Consistently with the hypothesis, TAZ failed to rescue transcription by p.N211S NKX2-1 on the lung-specific promoter SFTPB. Clinically, this is consistent with the presence of lung defects in patients with BLTS carrying this variant.
Our results support the strategy of simultaneous testing of NKX2-1 and WWTR1 (TAZ) in patients with suspected BLTS. Concurrent variants in WWTR1 (or PAX8) may contribute to the clinical presentation and understanding of genotype–phenotype correlations in BLTS. WWTR1 screening and in vitro rescue experiments may be particularly useful for fine-tuning pulmonary support and therapies. 35,36
To date, there are no known modifier genes for the neurological phenotypes of BLTS. Chorea is the most common feature, but its severity varies widely. Indeed, patients with only hypothyroidism and respiratory defects have been reported, 3,37,38 which may reflect some degree of transcriptional rescue at neuronal promoters. Brain-specific transcription partners or coactivators for NKX2-1 in striatal neurons or cortical interneurons may be the best candidates.
In conclusion, the p.N211S variant prevented functional rescue by TAZ despite preserving the C- and N-TADs of NKX2-1. This highlights the importance of investigating other nuclear factors that may profoundly affect the transcriptional effects of NKX2-1 mutants in BLTS.
Footnotes
Acknowledgments
The authors thank Mrs. Mercedes Tanarro for her technical contribution, the family participating in this study for their active collaboration, and Dr. Kenneth McCreath for assistance with editing.
Authors’ Contributions
B.V.: Conceptualization, investigation, and writing—original draft. C.C.-L.: Investigation, methodology, and writing—original draft. A.H.: Investigation. L.G.: Investigation. R.S.: Investigation. D.N.D.-B.: Investigation and writing—original draft. P.A.: Formal analysis. J.T.: Investigation. F.G.-S.: Investigation. M.S.: Investigation. A.D.-P.: Investigation. P.L.: Validation and writing—reviewing and editing. J.D.O.-E.: Validation and writing—reviewing and editing. P.S.: Investigation, validation, and writing—reviewing and editing. J.C.M.: Conceptualization, supervision, validation, and writing—reviewing and editing.
Author Disclosure Statement
The authors have declared that no conflicts of interest exist.
Funding Information
J.C.M. was supported by
Supplementary Material
Supplementary Data S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4