
Abstract
Outbreaks of West Nile virus (WNV) have occurred intermittently in regions around the Mediterranean coast, and the virus may have become established in Northern Italy and Romania, with reported intermittent outbreaks in Spain, Hungary, and France. WNV has also spread rapidly throughout the Americas since its introduction into New York in 1999. This capacity to emerge in new geographical locations and to spread rapidly together with the current increase in incidence of other flaviviruses such as tick-borne encephalitis virus, dengue virus, and Usutu virus has prompted us to design a novel pan-flavivirus RT-polymerase chain reaction for the purpose of surveillance for a range of flaviviruses. The assay utilizes degenerate primers targeting the flavivirus NS5 gene (RNA-dependent RNA polymerase) and detects a range of flaviviruses, including WNV. A small panel of WNV bird samples obtained from the United States has been shown to be detected using this assay. The amplicon generated is of sufficient size to provide sequence data to confirm the identity of the virus detected and undertake limited phylogenetic analysis. Testing using this assay has shown its ability to detect a range of tick-borne flaviviruses, particularly louping ill virus that is endemic in areas of the United Kingdom. The assay has been used to survey 160 bird samples and 1000 mosquito samples from the United Kingdom and found no evidence for WNV.
Introduction
To date, only one flavivirus is known to be endemic in the United Kingdom, louping ill virus (LIV). This virus is present within Ixodes ricinus populations in moorland areas of United Kingdom and is manifest as encephalitic disease of sheep (Gritsun et al. 2003). A number of reports have suggested serological evidence for West Nile and Usutu virus in the United Kingdom (Buckley et al. 2003, Buckley et al. 2006). However, these reports have not been confirmed by isolation of virus or evidence of disease in horses, avian species, or humans. Subsequent surveillance in birds between 2001 and 2006 failed to demonstrate any circulating WNV activity in British species (Phipps et al. 2008).
The ability to detect a number of flaviviruses in a single assay offers economies of scale and effort. As a result, a large number of attempts have been made to develop such assays using polymerase chain reaction (PCR) technology (Table 1). All but one has targeted the nonstructural protein genes of the flavivirus genome as these regions show less sequence variation. The majority have targeted the NS5 gene as this shows most conservation across the flavivirus genome. To maximize the number of flaviviruses detected, we have used degenerate bases within both flanking primers that produce an amplicon of approximately 200 bp to ensure efficient real-time amplification and detection. The assay developed has been assessed against a range of zoonotic flaviviruses and used to screen panels of mosquito, avian, and livestock samples, particularly for the presence of WNV.
Den, Dengue virus; WNV, West Nile virus; JEV, Japanese encephalitis virus; YFV, Yellow fever virus; SLE, St. Louis encephalitis virus; MVE, Murray Valley encephalitis virus; TBEV, tick-borne encephalitis virus.
Methods
Viruses/RNA
WNV (strain NY99), Sindbis virus, TBEV, Louping ill virus, and Usutu virus were provided by the Centre for Ecology and Hydrology, Oxford, United Kingdom. WNV (Goose Israel 1998) was provided by the Kimron Veterinary Institute, Israel. The panel of TBEV isolates from the Czech Republic was provided by Institute of Parasitology, University of South Bohemia, Czech Republic. RNA for Rift Valley fever virus was provided by the Health Protection Agency (HPA), United Kingdom. RNA for Dengue virus and Japanese encephalitis virus were provided by the University of Liverpool, United Kingdom. Louping ill isolates from the United Kingdom were submitted from Veterinary Laboratories Agency (VLA) regional laboratories at Penrith and Aberystwyth. Pooled mosquito samples were collected from locations within southern England and provided by the HPA, United Kingdom. RNA samples prepared from WNV infected bird organs were provided by the United States Geological Survey (USGS) National Wildlife Centre. Bird organ samples and ruminant serum samples were provided by the Veterinary Laboratories Agency regional network. RNA was extracted from mouse brain homogenates or cell lysates using TriZol (Invitrogen) following the manufacturer's protocol.
Flavivirus plaque assay
Briefly, a 10-fold dilution series of virus was prepared in serum-free Eagle's minimal essential medium (Earles salts) with penicillin/streptomycin/mycostatin (EMEM-WOS) in a 96-well plate, and transferred to a 12-well plate coated with Vero C1008 cells. After incubation at 37°C/5% CO2 for 1 h, 1.5 mL of warmed 1.5% carboxymethylcellulose in EMEM overlay was then added to each well, and the plates were further incubated at 37°C/5% CO2 for 4 days. Cells were fixed with 10% neutral buffered formalin for 3 h, and stained with 2.3% crystal violet (Sigma). Plaques were counted for each well, and plaque forming units per milliliter (PFU/mL) was calculated.
Primer design
Primers for detection of all flaviviruses were designed by alignment of 21 nonstructural protein 5 (NS5) sequences obtained from GenBank. Sequences of the following viruses were used: Gadgets Gully virus, Kyasanur Forest disease virus, Negishi virus, Omsk hemorrhagic fever virus (strain Kubrin), Powassan virus, Russian spring–summer encephalitis virus, louping ill virus, WNV, Dengue virus types 1 and 2, Usutu virus, and a number of TBEVs. The sequences of both sense and antisense primers, and their location on the WNV genome are given in Table 2. The amplicon from this primer pair is located toward the end of the NS5 coding sequence (Fig. 1) and produces an amplicon of the expected size of 203 bp. Extensive use was made of degenerate bases to broaden the ability of the primer pair to detect as wide a range of viruses as possible.
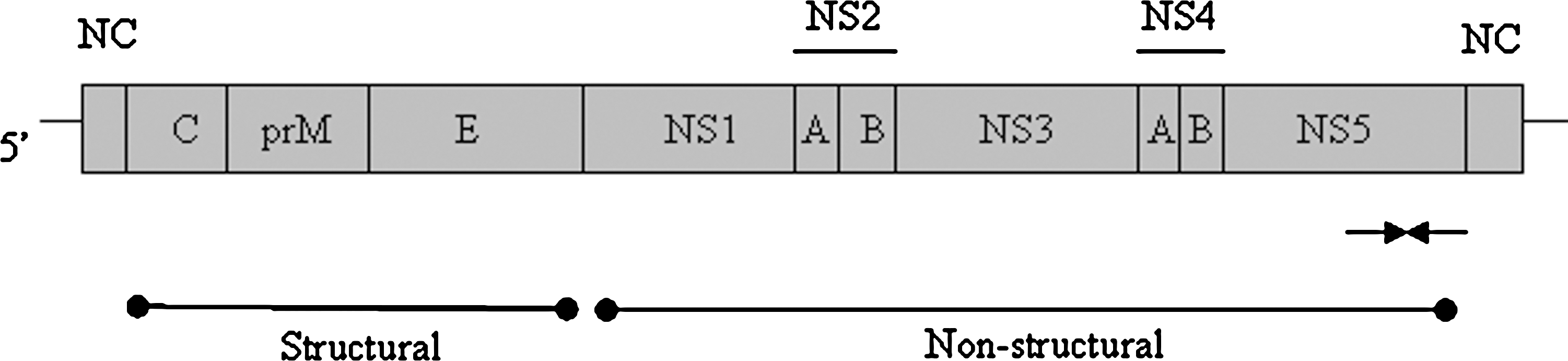
Schematic representation of the flavivirus genome showing the binding sites for primers Flavi-For and Flavi-Rev (arrows). NS represents nonstructural genes, and NC represents noncoding regions.
M = A+C; W = A+T; R = A+G; K = G+T; D = G+A+T; N = A+C+G+T.
Amplification method and product detection
Single-tube amplification was conducted using the QuantiTect kit (Qiagen), which includes Sybr green, with additional Quantiscript reverse transcriptase (Qiagen). One microliter of target RNA and 10 pmol of both primers were added to QuantiTect mastermix. The conditions used were reverse transcription at 50°C for 30 min followed by 15 min denaturation at 95°C followed by 40 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s. The reaction was completed by determination of the dissociation curve of all amplicons generated. All amplifications were performed using the MX3000p PCR system (Stratagene) and analyzed using MXPRO software (Stratagene). Positive amplifications show a fluorescence increase statistically higher than background expressed as a threshold cycle number (Ct).
Detection of WNV by TaqMan RT-PCR
WNV-specific primers and probe directed to the envelope-coding sequence (Lanciotti et al. 2000) were used to detect genomic RNA in organ samples obtained from infected U.S. birds (see above). The reactions were undertaken in a single-tube format using a modification of the lyssavirus assay described by Wakeley and co-workers (2005).
Sequencing/analysis (alignment & phylogenetic tree)
The identity of amplicons generated was confirmed by sequencing as previously described (Johnson et al. 2004). Sequences were edited using DNAstar package (Lasergene) and phylogenetic analysis was carried out using the PHYLIP program (version 3.5).
Results
Detection of arboviruses by pan-flavivirus primers
Assay specificity was assessed by analysis of a small panel of flaviviruses including two representative viral isolates of WNV. Real-time RT-PCR using the primer pair Flavi-For and Flavi-Rev successfully detected eight flaviviruses and failed to detect two mosquito-transmitted viruses unrelated to the Flavivirus genus (Table 3). Amplification curves (Fig. 2A) and dissociation curves (Fig. 2B) demonstrate the amplification of WNV isolates.
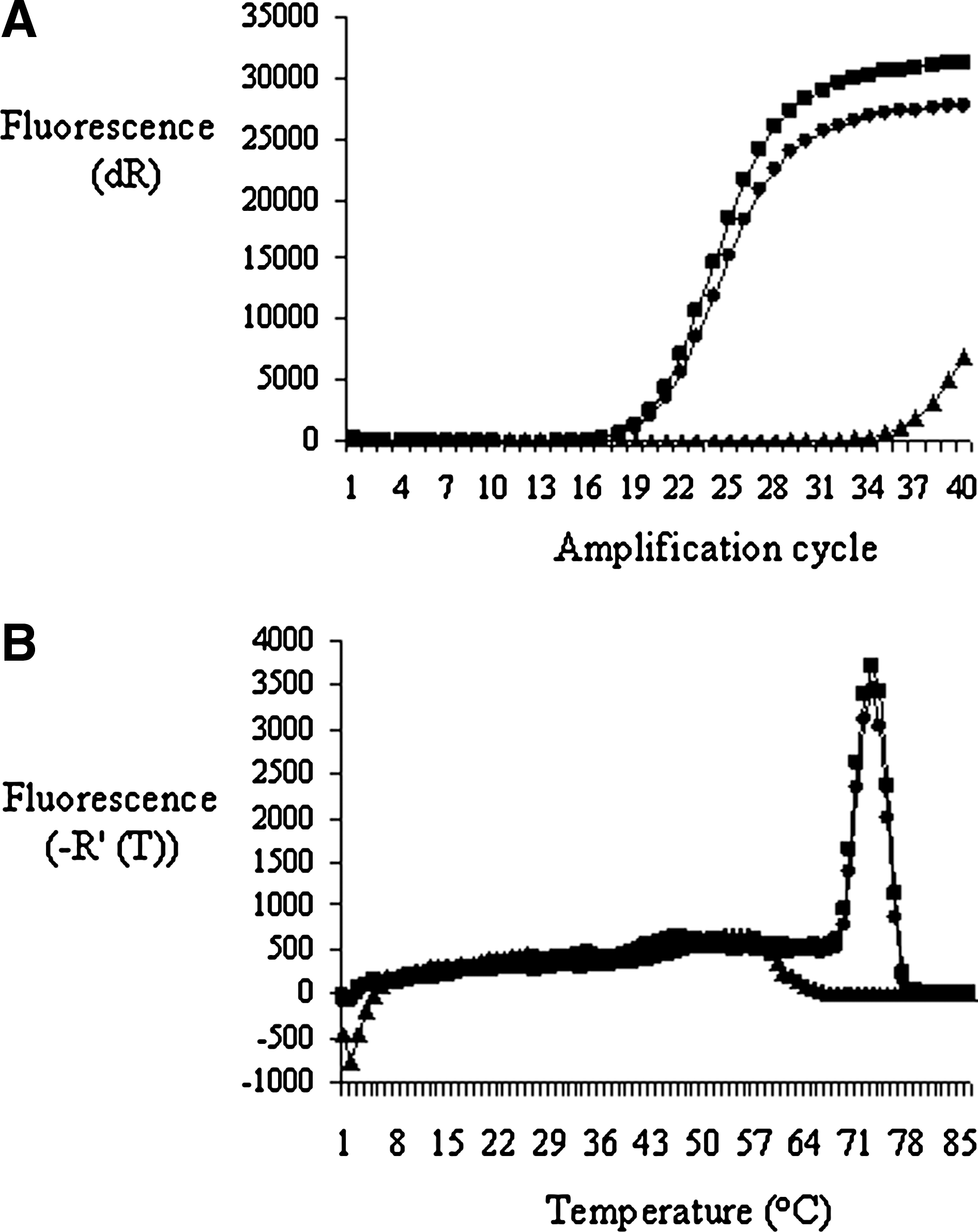
Detection of West Nile virus lineages 1 and 2 using pan-flavivirus primers. (
NA, not applicable.
Detection of WNV RNA samples by pan-flavivirus primers
A further panel of 20 WNV RNA samples prepared from bird organ samples (pooled kidney and spleen) in the United States was assessed using this assay (Table 4). In each case WNV was detected in each sample although with a higher Ct than a WNV-specific RT-PCR.
PCR, polymerase chain reaction.
Detection of TBEV by pan-flavivirus primers
To assess the ability of this assay to detect other flaviviruses in panels of samples, we used it to detect TBEV in a panel of 17 samples from the Czech Republic. Irrespective of the quantity of virus in each sample, as assessed by measurement of PFU, the assay detected flavivirus in each sample, including three samples where no PFU were detected (Table 5). The envelope protein of these isolates was sequenced to confirm their identity as TBEV isolates originating from the Czech Republic (data not shown).
PFU/mL, plaque forming units per milliliter.
Detection of louping ill virus field samples by pan-flavivirus primers
A key feature of this assay is its ability to detect the U.K. endemic flavivirus, Louping Ill virus (LIV). To assess this, samples were submitted by VLA regional laboratories in LIV endemic areas (Cumbria/Wales). RNA was extracted from spinal cord material from ewes showing neurological disease and in which disease had been confirmed by immunohistochemistry for LIV envelope protein. In three brain samples, LIV genomic RNA was detected in real-time with Ct values of 27.9, 23.7, and 38.8. Each amplicon had a dissociation temperature of 83.5 and could be detected at the expected size on an agarose gel (Fig. 3A). DNA sequencing produced sequences with strong homology to a laboratory strain of LIV but with some base substitutions reflecting their isolation from different geographical locations. This has been demonstrated in a larger study of regional variations of LIV (McGuire et al. 1998).
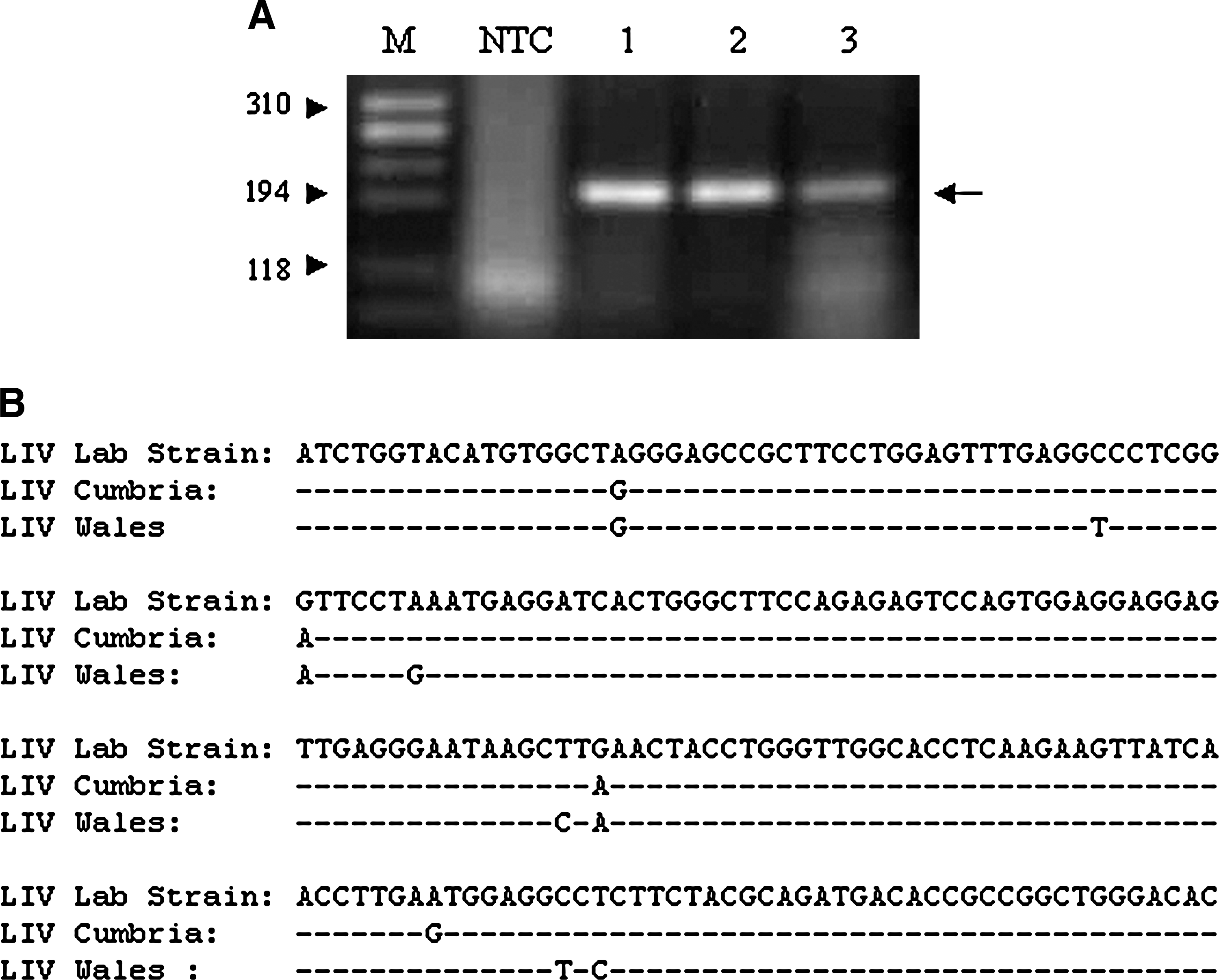
Detection of Louping Ill virus using pan-flavivirus primers. (
Surveillance for flaviviruses in U.K. avian and mammalian samples
This assay was used to assess three panels of samples obtained within the United Kingdom: 1000 mosquito samples collected and previously shown to be negative for WNV; one hundred and sixty bird brain samples collected from locations around England; and one hundred blood samples collected from sheep and cattle that were obtained for Anaplasma phagocytophila screening. In each case the assay returned negative results for flaviviruses.
Discussion
The ability of flaviviruses, particularly those transmitted by mosquitoes, to expand from their existing geographic range or to suddenly appear in new regions suggests that continued vigilance for these viruses is required (Gould et al. 2006). Global examples of flavivirus expansion include Dengue virus, yellow fever virus, and Japanese encephalitis virus. In a European setting, WNV and Usutu virus are examples of this phenomenon. Molecular techniques that detect a range of potential exotic flaviviruses should enhance surveillance. Factors such as the establishment of WNV in southern Europe, climate change, mosquito competence and density, wildlife population increases, and human activity may increase the probability that incursions of exotic flaviviruses will occur (Gale et al. 2009a, 2009b). Bird migration has also been reported as a means of WNV introduction (Malkinson et al. 2002) and many bird species migrate from Africa, across the Mediterranean to the United Kingdom. The assay described in this study is capable of detecting a range of flaviviruses using degenerate primers targeting the NS5 gene. The amplicon generated by this approach is small enough to enable efficient real-time detection of viruses through the incorporation of Sybr green, but of sufficient size to derive enough sequence data for further analysis of the PCR product as shown by the alignment of LIV sequences (Fig. 3). There is only one endemic flavivirus present in the United Kingdom, LIV, and this can be detected by the assay described in this study (Fig. 3). For detection of WNV, sensitivity appears to be the main limitation of this assay in direct comparison with WNV-specific TaqMan RT-PCR assays. This is illustrated in the difference in Ct values observed in a direct comparison between the two real-time RT-PCR assays used in this study (Table 4). Difference in threshold value is likely to result from the use of degenerate bases within the flanking primers and has been observed previously in the detection of WNV (Lanciotti et al. 2003). The main benefit of this assay is to provide wide coverage for a range of flaviviruses where a single virus cannot be specified. Sensitive, specific assays should be used where a single virus is suspected. The relatively small size of the amplicon suggests that inclusion of TaqMan probes would be one obvious development, an approach adopted by Dyer and co-workers (2007). An alternative might be more precise measurement of the dissociation curves generated from the amplicons of different viruses. Further validation of this assay will be required to assess its sensitivity for each flavivirus and to confirm that the sequence derived for each will provide sufficient phylogenetic discrimination. Preliminary sequence derived from this study identifies viruses to the species level, but further assessment is required to provide discrimination at the lineage level.
Two articles have reported serological evidence that West Nile has entered the United Kingdom (Buckley et al. 2003, 2006). Further surveillance has failed to detect this virus (Phipps et al. 2008); however, it is only through continued surveillance using detection methods such as those described in this study that will provide evidence for the presence or absence of flaviviruses within the United Kingdom. The sample surveys in this report have relatively low power and require larger sample sizes to enable a definitive assessment for the presence or absence for non-LIV flaviviruses in the United Kingdom. Such studies are being devised and it is only through continued surveillance that the methods and skills required to respond to such an outbreak are retained and refined by diagnostic laboratories.
Footnotes
Acknowledgments
The authors would like to thank the following for contributions of viruses and reagents: Libor Grubhopper and Daniel Ruzek (Institute of Parasitology, University of South Bohemia, Czech Republic), Mertyn Malkinson (Kimron Veterinary Institute, Israel), Ernie Gould (formerly Centre of Ecology and Hydrology, United Kingdom), Hon Ip (USGS National Wildlife Health Centre), Rebecca Mearns, Alexandra Schock (VLA), Richard Vipond, Jane Burton, Mark Outlaw (HPA, United Kingdom), Tom Solomon, and Sareen Galbraith (University of Liverpool, United Kingdom). The authors would also like to thank Kristy Murray (University of Texas School of Public Health) for useful discussions on WNV surveillance. Funding for this work was provided by Defra (Grant SE4106 and ED1600), EU FP7 coordinating action “ArboZooNet” International Network for Capacity Building for the Control of Emerging Viral Vector Borne Zoonotic Diseases and through an internal investment grant (TD0067).
Disclosure Statement
No competing financial interests exist.
©British Crown Copyright 2010.