
Abstract
Tick-borne diseases comprise a complex epidemiological and ecological network that connects the vectors, pathogens, and a group of host species. The aim of this study was to identify bacteria from the genus Rickettsia associated with ixodid ticks infesting camels and cows in Egypt. Ticks were collected from 6 different localities: Qina, Giza, Qalet El Nakhl, New Valley, El Arish, and Minufia, from July to October 2008. Species were identified using PCR, followed by sequencing. The gltA and rOmpA genes were used for the initial detection of Rickettsia spp. Further characterization of positive samples utilized primers targeting rOmpB, sca4, and intergenic spacers (mppA-purC, dksA-xerC, and rpmE-tRNAfMet ). Cows were infested with Hyalomma anatolicum excavatum and Boophilus annulatus. Camels were infested with Hyalomma dromedarii, H. impeltatum, and H. marginatum marginatum. Approximately 57.1% of H. dromedarii ticks collected from Qalet El Nakhl were infected with Rickettsia africae, exhibiting 99.1–100% identity to reference strains. Within H. impeltatum, 26.7% and 73.3% of ticks from El Arish were infected with R. africae and R. aeschlimannii, with 98.3–100% and 97.9–100% identity, respectively. Furthermore, 33.3% of H. marginatum marginatum ticks in Qalet El Nakhl were infected with the same two species as H. impeltatum, demonstrating 99.1–100% and 99.3–100% identity, respectively. By comparing percent identities and phylogenetic relationships, R. africae is identified for the first time in Egypt, in addition to R. aeschlimannii, which exhibits 100% identity with the Stavropol strain in GenBank. In conclusion, the obtained data underscore the medical and veterinary importance of tick-borne rickettsioses, which necessitate further investigation by authorities in Egypt. Moreover, additional characterization of these rickettsial isolates should be performed to designate their strains, using a polyphasic strategy combining genotypic and phenotypic tests, to facilitate their deposition in the rickettsial collection of the WHO and/or ATCC.
Introduction
The genus Rickettsia contains 25 officially-validated species and several as yet uncharacterized strains, 16 of which are recognized human pathogens, and another 2 are suspected to cause rickettsioses (Fourneir and Raoult 2009; Weinert et al. 2009). Worldwide, these zoonoses exhibit characteristic clinical features, including fever, headache, and occasional eschar formation at the site of the tick bite (Parola and Raoult 2001; Parola et al. 2005). Despite common ecological features and few phenotypic criteria, the classification and identification of Rickettsia have been based on differences in their epidemiology, serology, and intracellular growth characteristics (Philip et al. 1978). The main vectors of these bacteria are ticks (especially for spotted fever group rickettsiae, or SFG), which are also their reservoirs. These bacteria, which are transmitted among vectors via both transstadial and transovarial routes, may cause disease in invertebrate hosts in addition to vertebrate hosts, such as humans, domestic animals, birds, and wildlife (Raoult and Roux 1997; Anderson and Magnorelli 2008).
In Africa, little is known about the prevalence of these diseases. Cases of tick-borne rickettsioses have been regularly reported in North Africa since 1910 (Conor and Bruch 1910; Mediannikov et al. 2010). Rickettsia conorii was isolated in Tunisia in 1932 (Blanc and Caminopetros 1932). In 1992, Rickettsia africae was described, which appears to be distributed extensively throughout the continent, having been either isolated or detected by PCR in a number of African countries (Kelly et al. 1992). Rickettsia sibirica mongolotimonae, the agent of lymphangitis-associated rickettsiosis (LAR), has also been detected (Parola et al. 2001). Rickettsia slovaca, which causes tick-borne lymphadenopathy (TIBOLA) in Europe, has been found in Dermacentor marginatus ticks in Morocco by PCR (Sarih et al. 2008). Two Ixodes-associated rickettsiae, Rickettsia helvetica and Rickettsia monacensis, have also been detected in ticks in North Africa (Sarih et al. 2008; Dib et al. 2009). Rickettsia aeschlimannii was first isolated in Morocco in 1992 from the tick H. marginatum marginatum (Beati et al. 1997). Genotypically similar or identical strains have been reported in Egypt and Algeria (Bitam et al. 2006; Cazorla et al. 2008). Rickettsia massiliae was repeatedly detected in tick Rhipicephalus spp. in Algeria and Morocco in 1994 (Vitale et al. 2005). Many other SFG rickettsiae not previously associated with human diseases have been reported in ixodid ticks; in Africa, some species are genetically described and registered under Candidatus status (Matsumoto et al. 2006).
Mediterranean spotted fever, which is caused by Rickettsia conorii, is believed to have been enzootic for many years in Egypt and along the northern coast of Africa (Raoult and Roux 1997; Brown et al. 2005). Microscopic examination of the midguts and/or hemolymph of hard-bodied ticks, as well as serological, immunohistochemical, and molecular evidence, indicates that vectors, rodents, animals, and humans throughout Egypt are exposed to tick-borne rickettsial agents (McDade et al. 1973; Sixl et al. 1989; Botros et al. 1989,1995; Corwin et al. 1993; El Kady 1998). Rickettsiae of the typhus group (including Rickettsia typhi, the agent of murine typhus, and Rickettsia prowazekii, the agent of epidemic typhus) and the SFG (Rickettsia aeschlimannii, MC19 strain) (Ormsbee et al. 1968; Corwin et al. 1992; Lange et al. 1992; Reynolds et al. 2004) have been isolated from Rhipicephalus sanguineus and three Hyalomma spp. (H. dromedarii, H. impeltatum, and H. marginatum rufipes) ticks collected from Siwa Governorate, El Kharga Oasis, and Farghal village in Ismailia Governorate (Loftis et al. 2006b). Previous investigations demonstrated the possibility of a sampling bias in studies, which necessitates evaluating the incidence of unrecognized infections with R. prowazekii, R. typhi, R. conorii, R. aeschlimannii, and/or other unnamed Rickettsia spp. that have been detected in Egyptian arthropod vectors and animals (Soliman et al. 1989; Loftis et al. 2006a).
Over the last 15 years, detection of rickettsial DNA has dramatically increased the number of identified species (Raoult and Roux 1997). Multilocus sequence typing (MLST), which analyzes a minimum of 5 genes in combination, was developed for the identification of bacterial isolates and genotyping, and was subsequently proposed as a taxonomic tool for Rickettsia species (Stackebrandt et al. 2002). Genes encoding citrate synthase (gltA) (Roux et al. 1997), and the outer membrane protein A of the cell surface antigen family, rOmpA (sca0), which are only present in SFG rickettsiae (Roux et al. 1996; Fournier et al. 1998), outer membrane protein B, rOmpB (sca5) (Ching et al. 1990), and cell surface antigen 4, sca4 (gene D) (Sekeyova et al. 2001), have been used to rapidly and reliably differentiate members of the genus Rickettsia, either by analysis of restriction fragment length polymorphisms of PCR products (PCR-RFLP), or by direct sequence determination (Roux et al. 1997; Fournier et al. 1998; Roux and Raoult 2000; Sekeyova et al. 2001).
The present study aimed to identify and characterize species of Rickettsia associated with ixodid ticks, which were collected from different localities in Egypt, by sequencing PCR amplicons from bacterial DNA. Phylogenetic trees were constructed to illustrate the evolutionary relationship of Egyptian isolates to strains in GenBank.
Materials and Methods
Tick specimens
Ixodid ticks were collected from domestic camels (n=10) and cows (n=9) at the local animal trade market in each governorate. Collection areas included 6 localities: Qina, Giza, Qalet El Nakhl (Middle Sinai), New Valley, El Arish (North Sinai), and Minufia. Samples were collected from July to October 2008. Tick species were identified in the laboratory using the taxonomic keys of Hoogstraal (1956), Walker and associates (2003), and Estrada-Peña and colleagues (2004).
DNA extraction
Under sterile conditions, ticks were individually dissected and homogenized. Genomic DNA was extracted using the QIAamp® DNA tissue extraction kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. DNA from each tick was eluted in 200 μL of TE buffer and stored at −20°C until used for PCR.
Oligonucleotide design
PCR reactions were performed using the oligonucleotide sets listed in Table 1, which were purchased from GIBCO BRL (Life Technologies Sarl. B.P., Cergy Pontoise Cedex, France). Primer pairs recognizing genes encoding gltA and rOmpA, which are only present in SFG rickettsiae, were used for the initial detection of Rickettsia in total DNA (Roux et al. 1997; Fournier et al. 1998). Rickettsia-positive samples were further characterized using primers targeting rOmpB (Roux and Raoult 2000), sca4 (Sekeyova et al. 2001), and intergenic spacers (mppA, dksA, and rpmE) (Fournier et al. 2004).
PCR cycling parameters
Amplification reactions were performed on a Peltier model MJ Research PTC-200 thermal cycler (GMI, Inc., Ramsey, Minnesota) with the following conditions. DNA was initially denatured at 95°C for 15 min, followed by 40 cycles consisting of denaturation at 95°C for 1 min, annealing at the temperature indicated in Table 1 for 30 sec and extension at 72°C for 1 min. A final extension cycle at 72°C for 5 min was performed, and reactions were cooled to 15°C. The PCR reaction mixtures involved the use of 3 μL of extracted total tick DNA amplified in a 22 μL of reaction mixture containing 0.5 μL of each 100-mM primer, 2.5 μL of dNTP (dGTP, dATP, dTTP, and dCTP, 200 mM), 2.5 μL of PCR buffer 10×, 1.0 μL of MgCl2, 0.125 μL of Taq DNA polymerase, and 14.875 μL of twice-autoclaved sterile Milli-Q water. For each reaction, a negative control of sterile Milli-Q water was used. DNA isolated from R. africae or R. conorii was used as a positive control for all Rickettsia specimens investigated. PCR products were resolved by electrophoresis on a 1.5% agarose gel, then stained with ethidium bromide (15 μL/50 mL) in 0.5×TBE buffer for 20 min at 135 V, and were photographed and analyzed against the BenchTop pGEM® DNA Marker (Promega, Fitchburg, WI).
Sequencing of PCR products
Each amplicon was purified for sequencing using the QIAquick Spin PCR Purification kit (Qiagen, Courtaboeuf, France) according to the manufacturer's instructions. Sequencing reactions were performed with different sets of oligonucleotides (Tables 1 and 2), and the BigDye Terminator DNA sequencing kit (Perkin-Elmer, Waltham, MA), as described by the manufacturer. Each sequencing reaction was repeated at least three times in both the forward and reverse directions before being accepted for analysis.
Sequence analysis and construction of phylogenetic trees
Sequences of rickettsial gltA, rOmpA, rOmpB, and sca4 amplicons were aligned. The obtained sequences were assembled using ChromasPro 1.49 beta (Technelysium Pty. Ltd., Tewantin, QLD, Australia). Phylogenetic relationships between new Egyptian isolates and other reference strains published in GenBank were inferred using the BioEdit sequence alignment editor (v. 7.0.9.0). Two phylogenetic trees were constructed with the neighbor joining method (NJ) (Saitou and Nei 1987), and the unweighted pair group method with arithmetic mean (UPGMA) (Sneath and Sokal 1973; Dawyndt et al. 2006). The evolutionary distances were calculated by the maximum composite likelihood method (Tamura et al. 2004). The sequences were aligned using the program ClustalW 1.8® (Dessenet et al. 1990). Branches corresponding to partitions reproduced in less than 50% of bootstrap replicates were collapsed. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (100 replicates) is shown next to the branches (Felsenstein 1985,2004). Phylogenetic analyses were conducted in MEGA4 (Tamura et al. 2007).
Nucleotide sequence accession numbers
Percent sequence identity of Egyptian isolates with both R. africae and R. aeschlimannii reference strains were calculated (Table 3). Further analyses with other members of SFG rickettsiae were performed. The strains that were utilized for alignments, and the construction of phylogenetic trees, are indicated in Figures 3 –6.
Sequencing with rOmpB, sca4 and intergenic spacers (mppA, dksA, and rpmE) specified primers were applied for gltA and rOmpA genes PCR positive specimens.
Sequencing with sca4 specified primers were applied for Qalet El Nakhl H. dromedarii tick specimens only.
Rickettsia africae strain ESF-5 accession no. CP001612.1.
Rickettsia aeschlimannii strain Stavropol accession no. DQ235777.1.
Rickttsia aeschlimannii strain MC16 Rc-65 tandem repeat locus accession no. AY820043.1.
Rickttsia aeschlimannii strain MC16T rpmE-tRNAfMet accession no. DQ008255.1.
Rickttsia aeschlimannii strain MC16T mppA-purC accession no. DQ008293.
Results
Prevalence of intracellular bacteria in ixodid ticks
The incidence of intracellular bacteria in ixodid ticks (Fig. 1) collected from camels and cows was calculated for 6 localities in Egypt (Fig. 2). No Rickettsia spp. were detected among ticks collected from Qina, New Valley, Giza, or Minufia. However, H. anatolicum excavatum and H. dromedarii specimens from Qalet El Nakhl and El Arish, respectively, were uninfected. Overall, 100% of H. impeltatum ticks from El Arish, 57.1% of H. dromedarii, and 33.3% of H. marginatum marginatum tick specimens from Qalet El Nakhl were positive for Rickettsia spp. (Table 4).

Photos of ticks collected during the study. (

Map of Egypt showing the tick collection sites. Color images available online at
Partial amplification of the rOmpA gene and sequence analysis
Ticks infected with SFG Rickettsia spp. were characterized by analyzing the PCR products of the rOmpA gene. PCR using the specified primers on DNA isolated from ticks yielded fragments of 590 bp or 634 bp in length. Positive results were obtained for camel specimens only, specifically among H. dromedarii, H. marginatum marginatum, and H. impeltatum ticks collected from Qalet El Nakhl and El Arish (Table 4), whereas all cow specimens were negative. All remaining ticks collected from both animal hosts in the specified localities and classified as H. anatolicum excavatum and B. annulatus were negative by PCR using rOmpA gene primers (Table 4). Nucleotide sequence identities for the partial rOmpA gene amplicons ranged from 99.1–99.2% and 100% using BLASTN with the reference strains R. africae strain ESF-5 and R. aeschlimannii strain Stavropol, respectively (Table 3).
Amplification of the gltA gene and sequence analysis
Fragments of 852 bp to 1265 bp were obtained from the PCR amplification of Egyptian rickettsiae for the gltA gene. Sequence identities of 99.3–99.5% and 99.7–99.8% were obtained with the published sequence of R. africae strain ESF-5 and R. aeschlimannii strain Stavropol, respectively (Table 3).
Amplification of rOmpB (sca5) and sequence analysis
A sequence identity of 99.1–99.7% was obtained with the published sequence of R. africae strain ESF-5, whereas 99.3–99.8% identity was demonstrated with the published sequence of R. aeschlimannii strain Stavropol (Table 3).
Amplification of sca4 (gene D) and sequence analysis
Fragments of sca4 were amplified from Rickettsia-positive specimens and studied using three complementary PCRs (Table 1), spanning a total of 1277 bp. The sca4 sequence amplified is predicted to lie between bases 21 and 3050 for most Rickettsia spp. infecting ticks, according to the published sequence of the R. africae ESF-5 reference strain. The amplified open reading frames (ORFs) demonstrated 99.7% nucleotide sequence identity with R. africae (Table 3).
Amplification of mppA-purC, dksA-xerC and rpmE-tRNAfMet intergenic spacers and sequence analysis
Fragments of 155–197 bp, 164–292 bp, and 297–417 bp in length were obtained from the PCR amplification of Egyptian rickettsiae for the mppA-purC, dksA-xerC, and rpmE-tRNAfMet intergenic spacers, respectively. According to GenBank records, for each of the positive control strains, the following amplicon sizes were expected from PCRs using primers designed for the indicated intergenic sequences: a 160-bp amplicon for the mppA-purC sequence from both R. aeschlimannii MC16T and R. africae ESF-5 reference strains; 164-bp and 292-bp amplicons for the dksA-xerC sequence in R. aeschlimannii MC16T and R. africae ESF-5, respectively; and 416-bp and 337-bp amplicons for the rpmE-tRNAfMet sequence in R. aeschlimannii MC16T and R. africae ESF-5, respectively. BLAST analysis of the sequenced products revealed 99.5–100% and 97.9% sequence identities with R. africae ESF-5 and R. aeschlimannii MC16T, respectively, for the mppA-purC intergenic spacer (Table 3). The dksA-xerC intergenic spacer sequence exhibited 97.8%, 98.3%, and 100% identity with R. africae ESF-5, and 99.4–100% identity with R. aeschlimannii MC16T (Table 3). In the case of the rpmE-tRNAfMet intergenic sequences, identities of 99.7% with R. africae strain ESF-5 and 99.3–100% with R. aeschlimannii MC16T were demonstrated (Table 3).
GenBank accession numbers of Egyptian rickettsial isolates
All details of the Egyptian rickettsial isolates, including name, locality of collection, tick species, amplified sequence, and accession number for each record are listed in Tables 5 and 6. Eleven accession numbers were released for rOmpA (sca0) PCR amplicons. Additionally, 6 records were deposited in GenBank for rOmpB (sca5) amplicons, whereas for gltA, 10 records were deposited. The sca4 gene (gene D) has only one record. Finally, intergenic spacers comprise 14 records: 4 for mppA-purC sequences, 5 for dksA-xerC fragments, and 5 records for rpmE-tRNAfMet PCR products.
Construction of phylogenetic trees
Phylogenetic relationships were inferred from the aligned sequences of the amplified genes (Figs. 3 –6). Similar topologies were predicted by both NJ (data not shown) and UPGMA (Figs. 3 –6), which confirms the evolutionary relationship of the Egyptian isolates to SFG reference strains included in the phylogenetic construction. Therefore, only UPGMA trees are presented (Figs. 3 –6) and discussed.

Phylogenetic tree based on the sequences of PCR amplicons from the outer membrane protein A gene (rOmpA) of SFG rickettsiae detected in H. dromedarii, H. impeltatum, and H. marginatum marginatum with the UPGMA method (MEGA4 software). Two different topologies were produced with the nearly identical reference strains used in this study as positive controls: R. africae ESF-5 strain and R. aeschlimannii strain Stavropol (PCR, polymerase chain reaction; UPGMA, unweighted pair group method with arithmetic mean).

Phylogenetic tree based on the sequences of PCR amplicons of the cell surface antigen 4 gene (sca4) in H. dromedarii (

Phylogenetic tree based on the sequences of PCR amplicons of the outer membrane protein B gene (rOmpB) of SFG rickettsiae detected in H. marginatum marginatum (

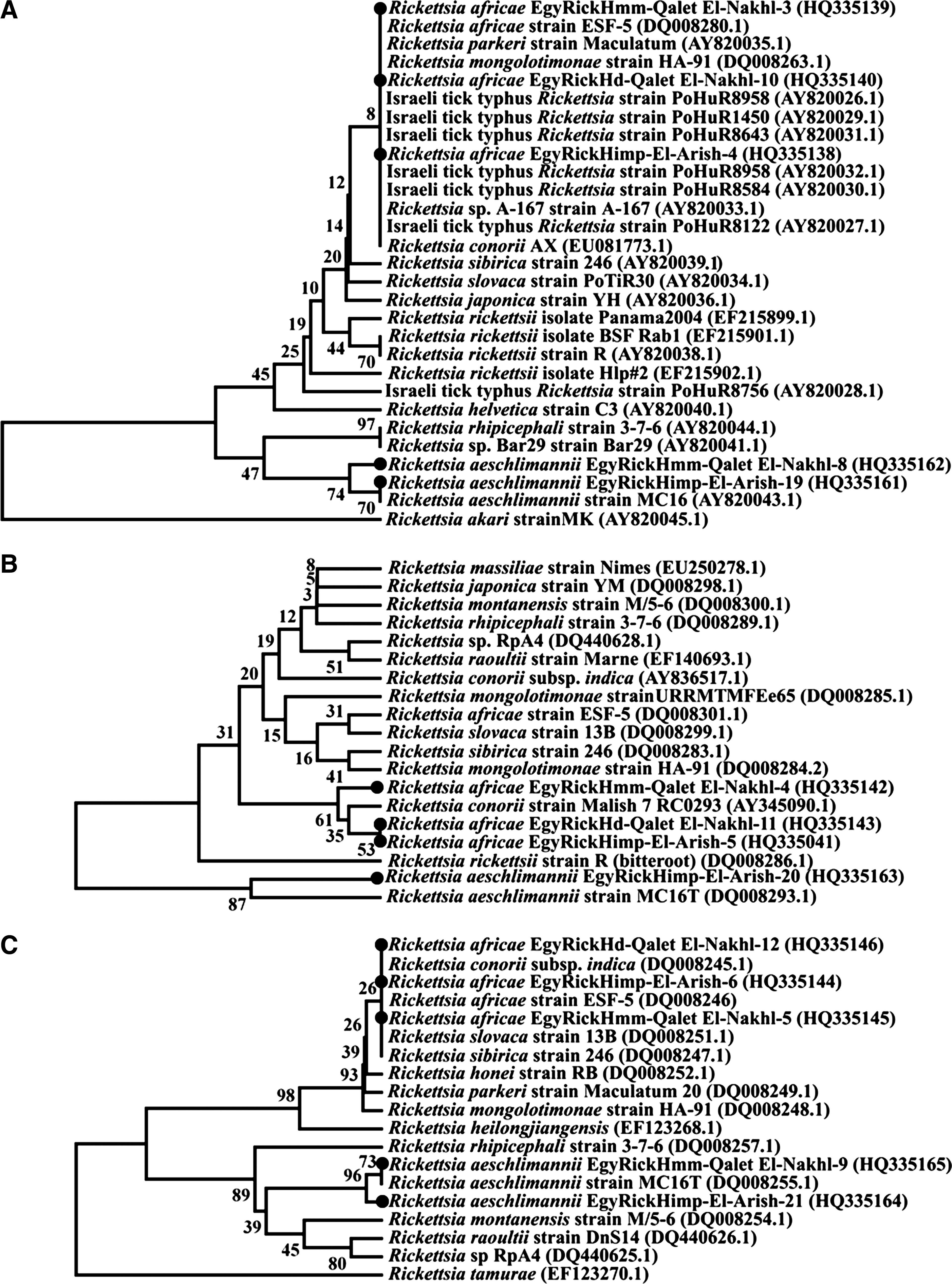
Phylogenetic tree based on the sequences of PCR amplicons of intergenic spacers (dksA-xerC, mppA-purC, and rpmE-tRNAfMet ) of SFG rickettsiae detected in H. dromedarii, H. impeltatum, and H. marginatum marginatum with the UPGMA method (MEGA4 software). Two different topologies were produced with the nearly identical reference strains used in this study as positive controls: R. africae ESF-5 strain and R. aeschlimannii strain MC16. A-dksA-xerC intergenic spacer aligned with R. africae strain ESF-5 and R. aeschlimannii strain MC16. B-mppA-purC intergenic spacer aligned with R. africae strain ESF-5 and R. aeschlimannii strain MC16. C-rpmE-tRNAfMet intergenic spacer aligned with R. africae strain ESF-5 and R. aeschlimannii strain MC16 (PCR, polymerase chain reaction; UPGMA, unweighted pair group method with arithmetic mean; SFG, spotted fever group Rickettsiae).
Discussion
Rickettsia spp. have been detected in a wide range of hosts, suggesting that they are more common than had previously been suspected (Weinert et al. 2009). The belief that the ecological characteristics of ticks as vectors influence the epidemiology and clinical aspects of tick-borne diseases informed our choice of the governorates included in this study. These ecological considerations in the transmission of rickettsiae could explain the negative results of PCRs on DNA from B. annulatus and H. anatolicum excavatum, and that the Giza, New Valley, Qina, and Minufia governorates were Rickettsia-negative despite the prevalence of animal trading and the presence of rodents along both sides of the Nile River (Fig. 2 and Table 4). These results illustrate the necessity for additional studies. Hence, we still do not have a complete understanding of how rickettsiae are maintained within vector populations or how they are transmitted horizontally between vector species (Parola and Raoult 2001). A better understanding of these dynamic processes should be the subject of future research, with detailed studies of representatives from the different groups of animals, vectors, rodents, and humans that reside in these localities.
The results reported here are in agreement with those that previously demonstrated the existence of R. aeschlimannii in Egypt. Here, the PCR-amplified rickettsial sequences exhibited greater than 99% identity to the Stavropol and MC16 strains (Figs. 3 –6 and Tables 4 –6). R. aeschlimannii is transmitted by H. impeltatum and H. marginatum marginatum in two districts of the Sinai Peninsula: El Arish and Qalet El Nakhl, which are near Ismailia which is at the western entrance of the Sinai, along with the Suez Canal, where R. aeschlimannii was first detected (Loftis et al. 2006a and b) (Fig. 2). The repeated, precise clusters of the Egyptian rickettsial isolates with R. aeschlimannii strains are prominent in the phylogenic trees inferred from the sequences of the rOmpA (Fig. 3) and rOmpB (Fig. 5A and B) genes, as well as the three intergenic spacers: dksA-xerC (Fig. 6A), mppA-purC (Fig. 6B), and rpmE-tRNAfMet (Fig. 6C), when aligned with similar GenBank records. Despite the fact that these topologies, combined with previous results, demonstrate the endemicity of R. aeschlimannii in Egypt, they do not indicate the directionality of rickettsial transmission, whether from the Sinai to the delta and western Egypt or vice versa; thus further investigation is needed.
The topology of the phylogenies inferred by the analysis of the PCR-amplified rickettsial sequences of certain divergent genes, including gltA, rOmpA, and rOmpB (Roux and Raoult 1995; Roux et al. 1997; Fournier et al. 1998; Roux and Raoult 2000; Sekeyova et al. 2001), enabled the detection of R. africae isolates for the first time in Egypt; these strains approach 100% identity to strain ESF-5, the causative agent of African tick bite fever, which is widely distributed throughout the African continent (Roux and Raoult 2000). H. dromedarii, H. impeltatum, and H. marginatum marginatum phylogenies are supported by the topology inferred from non-coding intergenic spacers, which are better able to distinguish among rickettsiae at the strain level, and to trace isolates of a single rickettsial species based on the assumption that intergenic regions experience less evolutionary pressure than coding sequences (Fournier et al. 2004). However, the obtained spacer clusters illustrate a relationship between Egyptian isolates and other rickettsial species, such as R. africae with R. conorii, R. sibirica, R. slovaca, R. mongolotimonae, and Israeli tick typhus (dksA-xerC [Fig. 6A], mppA-purC [Fig. 6B], and rpmE-tRNAfMet [Fig. 6C]), illustrating the need for further characterization of these amplicons using other genome constituents. Tick-borne rickettsioses play a very important role in public health. A recent worldwide report demonstrated a 5.6% incidence of rickettsial infection in a group of travelers who developed acute febrile infections after returning from Africa. After malaria, rickettsiae are among the most frequently identified etiologies for systemic febrile illness among travelers (Freedman et al. 2006; Jensenius et al. 2009).
The fact that tick-associated rickettsiae are characterized by limited and specific geographical distribution (Anderson and Magnorelli 2008) may explain the detection of R. africae solely in H. dromedarii collected from Qalet El Nakhl (Middle Sinai Peninsula) and not from El Arish (North Sinai Peninsula). Furthermore, the distance and lack of contact between the targeted locations in this study with urban Siwa Governorate and El Kharga Oasis (Fig. 2), which were surveyed previously (Loftis et al. 2006b), may explain the variations in the percent identities between R. aeschlimannii strains seen in the two studies. However, the existence of mixed rickettsial infections (R. aeschlimannii and R. africae), as detected in H. marginatum marginatum and H. impeltatum (Tables 5 and 6), may be relevant to the recent discovery of plasmids in the genus Rickettsia, which could affect horizontal gene transfer and recombination between rickettsial strains (Weinert et al. 2009). This finding has important implications for the evolution of rickettsiosis because genes can sweep through different genetic backgrounds, thereby altering bacterial pathogenicity and transmission capacity. However, the least studied role of rodents (Loftis et al. 2006a) that reside throughout the Sinai desert in maintaining a pool for these pathovars is unfortunate; comparing vertebrates as reservoirs with susceptible hosts and vectors may help to elucidate the mechanisms of pathogenicity, transmission, and virulence of rickettsiosis.
All known vertebrate-associated rickettsiae that are transmitted by arthropods as part of their life cycle do not normally infect humans during their natural cycles between arthropod and vertebrate hosts (Roux and Raoult 2000). In addition, when rickettsiae are transmitted efficiently, both transstadially and transovarially, ticks will serve as reservoirs of the bacteria, and the distribution of rickettsiosis will be identical to that of its tick host, which illustrates the danger of the live animal trade introducing ticks into different habitats (Parola and Raoult 2001). These remarks should highlight the importance of considering these rickettsial isolates as strains of unknown future pathogenicity for both humans and animals, rather than nonpathogenic, because they could be associated with arthropods that feed on humans and travelers to Egypt, as well as drift according to their rodent and mammalian host movements (Parola and Raoult 2001; Pretorius and Birtles 2002). The role of uncontrolled rodent populations in pathogen exchange and the effects of these bacteria on their arthropod hosts should be fully understood by entomologists.
In conclusion, the use of PCR and sequencing methods for the identification of SFG rickettsiae in ticks has led to new questions regarding the geographical distribution of tick-borne rickettsiae and the association between rickettsiae and ticks in Egypt, as well as along the northern coast of Africa. Genotypes similar to R. africae were found in H. dromedarii, H. marginatum marginatum, and H. impeltatum, and genotypes similar to R. aeschlimannii were found in H. marginatum marginatum and H. impeltatum. Further characterization of Egyptian rickettsial isolates to designate their type strains using a polyphasic strategy combining genotypic and phenotypic tests will facilitate their deposition into the rickettsial collection of the WHO and/or ATCC.
Footnotes
Author Disclosure Statement
No competing financial interests exist.