
Abstract
Antibodies against non-structural protein 1 (NS1) are considered to be the most reliable indicator of a present or past infection by West Nile virus (WNV) in animals. In this study, an in-house competitive enzyme-linked immunosorbent assay (NS1-cELISA) utilizing baculovirus-expressed NS1 and monoclonal antibodies against NS1 was established for the detection of antibody responses to NS1 in WNV-infected animals. The assay was validated by the simultaneous detection of early antibody responses to NS1 and the structural envelope protein in animals infected with WNV, or inoculated with inactivated WNV. NS1-cELISA detected WNV antibodies at 6 days post-infection (dpi) in a WNV-infected rabbit (percent inhibition [PI] value of 84.0), and at 10 dpi in a WNV-infected chicken (PI value of 67.0). The NS1-cELISA was able to detect WNV antibodies in sera from all WNV-infected rabbits at 10 dpi (PI value of 79.2±18.0), and from three of four WNV-infected chickens at 14 dpi (PI value of 73.7±22.8). The results of this study demonstrate that the antibody response to NS1 is similar to that against envelope protein in WNV-infected rabbits and chickens, whereas animals inoculated with inactivated WNV develop antibody responses only to the envelope protein but not to NS1. The NS1-cELISA developed here has the potential to be a useful tool for monitoring WNV circulation (i.e., the prevalence of specific antibodies against WNV NS1), by assaying serum samples from regions in which an inactivated vaccine control strategy has been implemented.
Introduction
When animals are immunized with an inactivated vaccine, they mount antibody responses only against the structural proteins of the virus; however, when animals become infected, antibodies against non-structural proteins (NSPs) such as viral polymerases and proteases also develop because the virus replicates inside the host (Sutmoller et al. 2003). The detection of NS1 antibody in serum indicates that an animal has come into contact with wild-type virus. Such tests are especially important in the vaccination scenario because no other methods are suitable for the large-scale evaluation of the effectiveness of disease-control measures adopted in response to an outbreak. For these reasons, NSP antibody-detection methods have been extensively investigated in recent years (Rodriguez, et al. 1994; Lubroth and Brown 1995; Sorensen et al. 1998; Bergmann et al. 2000; Brocchi et al. 2003; Robiolo et al. 2006), and several kits are commercially available. In addition, regarding arboviruses such as bluetongue virus and African horse sickness virus, non-structural proteins have been investigated, and their potential as markers for differentiating infected animals from vaccinated animals has been demonstrated in previous studies (Bougrine et al. 1998; Barros et al. 2009). However, the use of an NSP ELISA suitable for differentiating WNV-infected animals from vaccinated animals has not been reported, and validated test kits are not yet commercially available.
In this study, we sought to develop and validate a competitive ELISA (NS1-cELISA) using baculovirus-expressed NS1 protein as the antigen of interest and monoclonal antibodies against NS1 for the differentiation of WNV-infected animals from vaccinated animals.
Materials and Methods
WNV culture and inactivation
WNV strains (strain NY385-99 [lineage I, ATCC VR-1507] and strain B956 [lineage II, ATCC VR-1501]) were obtained from the American Type Culture Collection (ATCC; Manassas, VA). The JEV strain Anyang300 (Yang et al. 2005) was also used in this study. Viruses were grown in Vero cells (ATCC CCL-81). WNV manipulations were performed in a BioSafety Level 3 (BSL-3) containment research laboratory at the National Veterinary Research and Quarantine Service (NVRQS; Anyang, the Republic of Korea) in accordance with the regulations of the Korean government. For the titration of WNV infectivity, a plaque assay was performed according to previously described methods (Payne et al. 2006).
The virus culture supernatant was clarified by treatment with protamine sulfate (0.8% w/v; Merck, Rahway, NJ) and centrifugation at 10,000×g. The virus was then concentrated by polyethylene glycol precipitation and inactivated by incubation with formaldehyde. To this end, formalin (37% formaldehyde; Merck) was diluted 1:40 in phosphate-buffered saline (PBS), and the pH of the solution was adjusted to 7.2 with 0.1 N NaOH. The diluted formalin was added drop-wise to the virus suspension. A final formaldehyde concentration of 10 mmol was used to inactivate the virus at 4°C, and this mixture was incubated for 30 days with regular shaking. A final formaldehyde concentration of 5 mmol/L was used for virus inactivation at 22°C, and this mixture was incubated for 10 days with regular shaking. The inactivated virus preparation was dialyzed against PBS to remove the formaldehyde and was tested for virus infectivity by inoculation of Vero cell monolayers. To prepare the inocula, the inactivated virus preparation was mixed with an equal volume of PBS and incubated at room temperature for 30 min.
Challenge and vaccination of animals
Rabbits with an average body weight of 2.0 kg were randomly allotted into three groups of six animals each, and 4-week-old chickens were also randomly allotted into three groups of four animals. Animal groups were injected IM with one of the following: 106 PFU of Vero cell-cultured WNV virus diluted in PBS, formalin-inactivated WNV mixed with PBS, or PBS only as a negative control. Serum samples from the inoculated animals were collected for the next 3 weeks. Inoculations of animals with live virus were performed in a BSL-3 containment research laboratory at the NVRQS in accordance with the regulations of the government of the Republic of Korea.
Field and reference serum samples
A total of 187 negative serum samples that were confirmed by running in-parallel plaque-reduction neutralization tests (PRNTs) for detecting WNV- or JEV-neutralizing antibodies in horse sera were used to determine a cut-off value. In the same manner, 160 naïve chicken and 24 naïve rabbit serum samples that had been confirmed as negative for viral infection by PRNT were also included to determine a cut-off value for the multi-species NS1-cELISA. To evaluate the NS-cELISA, the following reference equine and avian control serum samples were obtained from the Reference Laboratory of the World Animal Health Organization at the United States Department of Agriculture and the National Veterinary Services Laboratories (NVSL, Ames, IA), and BioReliance (Invitrogen Bioservices, Rockville, MD): W. NILE EQ AS VN336EDV0003BP192, WESTNILE EQ AS immunoglobulin M (IgM) 330EDV0201 Low Pos H351, W.N EQ AS IgM 330EDV 0202 Low Pos H354, WESTNILE EQ AS IgM 335EDV0002, WESTNILE EQ AS IgM 335EDV0003, Normal EQ Sera 305EDV0201 IgM, Normal EQ Sera 306EDV0201VN, Normal EQ Sera 306EDV9801, and High Positive/Low Positive/Negative WNV Avian Antibody set (cat. 1-014). These sera were used to validate the NS-cELISA in this study.
Expression and purification of recombinant NS1 protein
Plasmid construction and expression and purification of WNV NS1 from insect cells were performed as described by Chung and associates (Chung et al. 2006). The last 72 nucleotides of WNV E (an endogenous signal sequence), and the entire NS1 gene from the NY385-99 strain were amplified by high-fidelity PCR. As shown in Figure 1A, the PCR product was digested and inserted into the BamHI and XbaI sites of a baculovirus shuttle vector (pFastBac1; Invitrogen Corp., Carlsbad, CA) and sequenced. Soluble NS1 protein was generated using a commercially available baculovirus expression system (Bac-to-Bac system; Invitrogen). Three days after the baculovirus infection of serum-free adapted SF9 insect cells at a multiplicity of infection of 0.25, the supernatant was harvested, filtered, buffer-exchanged into 10% glycerol, 300 mM NaCl, and 25 mM Tris-HCl (pH 7.5), and then concentrated and purified by nickel affinity chromatography according to the manufacturer's instructions (Invitrogen). Immunoblotting was then performed by standard techniques using a rabbit polyclonal antibody (which is described below) against the NS1 protein.
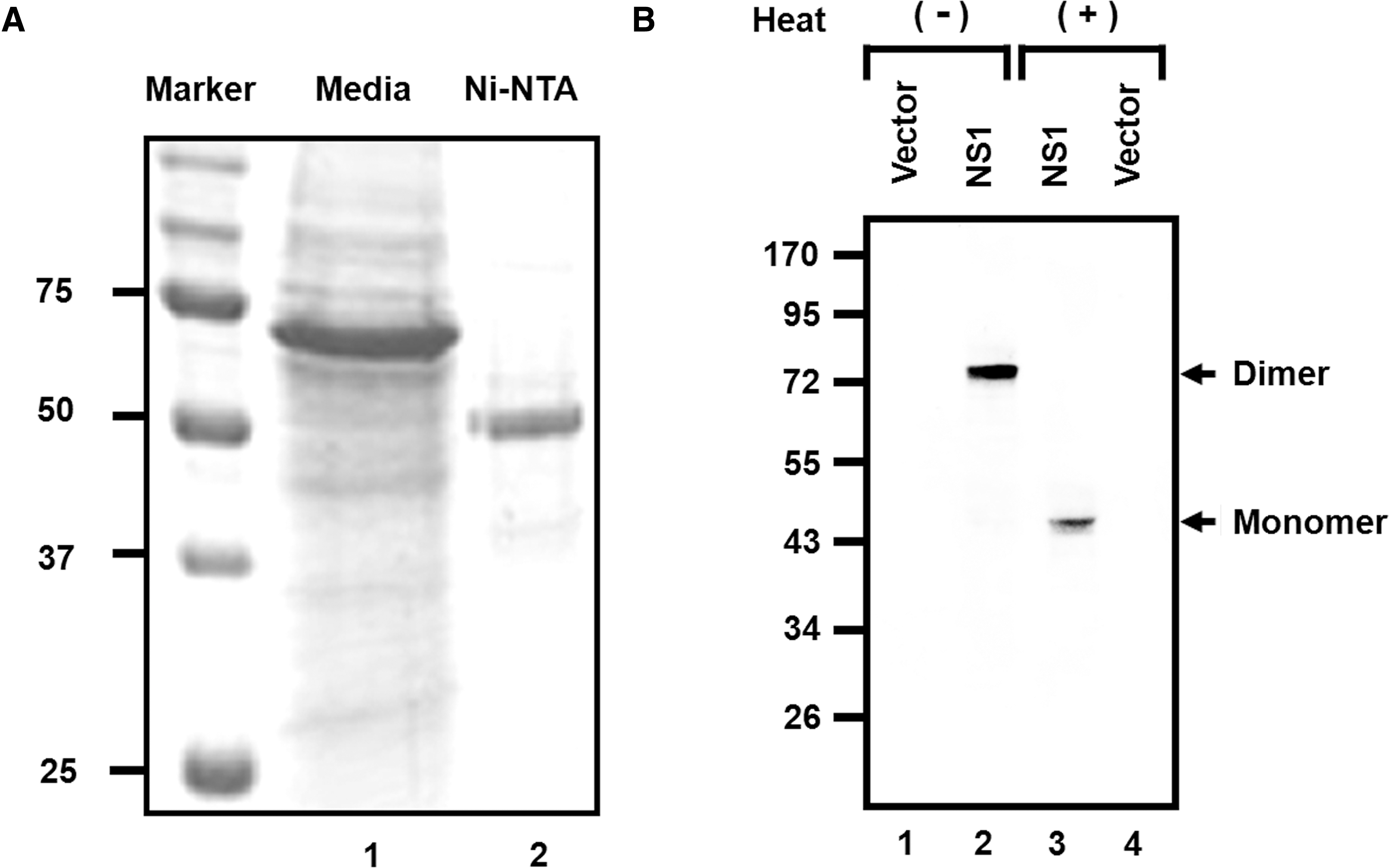
Biochemical characterization of West Nile virus (WNV) non-structural protein 1 (NS1). (
Generation of monoclonal and polyclonal antibodies against NS1
BALB/c mice were primed and boosted twice at 3-week intervals with insect cell-generated, purified, recombinant WNV NS1 protein complexed with Freund's adjuvant (Sigma-Aldrich, St. Louis, MO). For immunization, 200 μg of purified NS1 was injected with the same volume of incomplete Freund's adjuvant into the footpads of BALB/c mice. To generate hybridomas, spleen cells were harvested 3 days after the intravenous boost and fused to SP2/O myeloma cells using polyethylene glycol. Hybridoma cells were screened by indirect ELISA (see the section on indirect ELISA, below) using purified WNV NS1 protein as a coating antigen, followed by Western blotting. The clones were also screened by an immunofluorescence assay (IFA) using Vero cells infected with WNV lineages I (strain NY385-99) and II (strain B956), and Arbovirus Screen (Non-USA and USA) Slides (Panbio Limited, Windsor, Queensland, Australia) to investigate their cross-reactivity. The monoclonal antibodies were isotyped using the Mouse Monoclonal Antibody Isotyping Kit (Roche Applied Science, Indianapolis, IN), and purified using the ImmunoPure IgG and IgM purification kit (Thermo Fisher Scientific, Waltham, MA).
Preparation of polyclonal antibody
Male white rabbits were injected IM with 100 μg of recombinant NS1 protein emulsified with Freund's complete adjuvant (Sigma-Aldrich), followed by three booster doses of 150 μg of protein emulsified with incomplete Freund's adjuvant (Sigma-Aldrich) administered at intervals of 7 days. The animals were bled 7 days after the last injection, and antiserum was separated from the blood.
ELISA procedures
Indirect ELISA
Purified WNV NS1 protein generated from insect cells at 1.5 μg/100 μL per well was adsorbed overnight at 4°C on Maxisorp microtiter plates (Nalge Nunc International, Rochester, NY) to select the most suitable monoclonal antibody clones. Nonspecific binding was blocked after incubation with blocking buffer (0.05% PBS, Tween 20, 1% bovine serum albumin, and 3% equine serum) for 1 h at 37°C. The plates were then incubated with monoclonal antibodies against WNV NS1 (50 μg/mL) for 1 h at 37°C. After extensive washing, the plates were incubated serially with biotin-conjugated goat anti-mouse IgG and horseradish peroxidase-conjugated streptavidin (Sigma-Aldrich) at 37°C, and developed after the addition of 3,3′,5,5′-tetramethyl-benzidine substrate.
Competitive binding ELISA
The design of the NS1-cELISA was based on competitive binding of the anti-NS1 monoclonal antibodies b2NS14 and eNS02 and serum WNV antibodies from infected animals. Briefly, microplates sensitized with eNS02 monoclonal antibody were serially incubated with NS1 protein, test samples, the peroxidase-conjugated monoclonal antibody b2NS14, and the o-phenylenediamine dihydrochloride substrate (Sigma-Aldrich) for the colorimetric detection of horseradish peroxidase activity. The amounts of each reagent used in the NS1-cELISA were optimized by checkerboard titration. The optimal dilution of the peroxidase-conjugated monoclonal antibody b2NS14 was 1:500 (1.0 μg/mL), which had an optical density (OD) of 1.42±0.13, corresponding to 75% of the maximum absorbance at 492 nm. Dilutions (1:10) of control equine sera (strongly and weakly positive control sera and negative control sera from the NVSL), and a 1:500 dilution of peroxidase-conjugated b2NS14 monoclonal antibody optimally differentiated positive sera from normal negative sera. Using the conditions above described, the mean PI values for strongly positive (1:5 dilution in negative serum), weakly positive (1:50 dilution in negative serum), and negative rabbit sera were 94%, 58%, and 4.9%, respectively.
ELISA for NS1 antibody detection
Maxisorp ELISA plates were coated with the monoclonal antibody eNS02 (2.8 μg/mL) in 0.01 mol/L PBS (pH 7.4), 50 μL/well, and then incubated for 1 h at 37°C. The plates were washed three times with 0.01 mol/L PBS containing 0.05% Tween 20 and then incubated with 50 μL of the recombinant NS1 antigen, which corresponds to approximately 3.6 μg/mL of purified protein (as described above), in blocking buffer (0.01 mol/L PBS containing 0.05% Tween 20 and 5% skim milk) for 1 h at 37°C. After three washes, the plates were incubated for 40 min at 37°C with 50 μL of a mixture containing equal volumes of peroxidase-conjugated b2NS14 monoclonal antibody and serial dilutions of test sera. Positive and negative reference control panel sera that were obtained from NVSL and BioReliance (Invitrogen Bioservices) were included and tested to validate the NS-cELISA. After the washes, the plates were incubated for 10 min with o-phenylenediamine dihydrochloride substrate in 0.05 mol/L phosphate-citrate buffer (pH 5.0) containing 0.015% hydrogen peroxide. The colorimetric reaction was stopped by adding 50 μL of 1.25 mol/L sulfuric acid to each well. The OD was measured at a wavelength of 492 nm, and was converted to PI of monoclonal antibody binding by competition with serum antibodies using the following formula: PI=[1 − (OD of serum-monoclonal antibody mixture/OD of monoclonal antibody alone)]×100. To compare antibody responses to the structural envelope protein (E), serum samples were simultaneously tested using an in-house IgG ELISA (NT-ELISA, an enzyme-linked immunosorbent assay using 5E8 neutralizing monoclonal antibody [NVRQS, Anyang, the Republic of Korea]), according to the method described by Choi and colleagues (Choi et al. 2007).
Results
WNV NS1 expression and production of monoclonal WNV NS1 antibodies
Soluble WNV NS1 was harvested from baculovirus-infected SF9 insect cells and purified to homogeneity after sequential nickel affinity, size exclusion, and Mono Q fast protein liquid chromatography (Amersham Biosciences, Piscataway, NJ). Dimerization of WNV NS1 was detected after SDS-PAGE and Coomassie staining, and the identity of this protein was confirmed as WNV NS1 by Western blotting with a monoclonal antibody against WNV NS1 (Fig. 1). After screening more than 500 hybridomas, b2NS14 and eNS02 were selected for use in the sandwich ELISA from among 14 collected clones based on their specificity, antibody productivity, and cell growth rates (Table 1).
Competitive enzyme-linked immunosorbent assay using recombinant NS1 protein as a coating antigen.
Immunofluorescence assay (IFA) for the specificity of antibodies against Vero cells infected with WNV lineage I (strain NY385-99) and II (strain B956).
Arbovirus Screen (non-USA and USA) Slides (Panbio Limited) were used to evaluate the flavivirus antigenic specificity of the monoclonal antibodies. The specificities of the reactions are described.
Western blotting (WB) using recombinant NS1 protein.
JEV, Japanese encephalitis virus; SLEV, St. Louis encephalitis virus.
Cut-off value determination
The mean inhibition values of 160 naïve chicken, 187 naïve horse, and 24 naïve rabbit sera ranged from 9.0−11.5%, and no significant differences were observed between groups (p>0.05). Naïve serum samples were confirmed as negative for both WNV and JEV infection by PRNT as described above. We therefore used all groups of sera to determine the cut-off value for our competitive ELISA. These sera showed inhibition of b2NS14 binding ranging from 0.0−29.3%, with a mean of 8.8% and a standard deviation (SD) of 10.3%. We tentatively determined a cut-off value of 40.0%, which is approximately equal to the mean PI value of the negative sera plus three SD, to differentiate samples positive for WNV-NS1 antibodies from negative samples. Theoretically, the probability of a negative sample having a PI value greater than this cut-off value was calculated to be 0.16%.
Evaluation of the NS1-cELISA for the detection of WNV antibodies
The analysis of the performance of the NS1-cELISA at detecting antibodies against WNV NS1 in experimentally-infected chickens and rabbits is summarized in Figure 2. NS1-cELISA detected WNV antibodies at 6 dpi in a WNV-infected rabbit (PI value of 84.0; Fig. 2A), and at 10 dpi in a WNV-infected chicken (PI value of 67.0; Fig. 2C). The NS1-cELISA was able to detect WNV antibodies in sera from all WNV-infected rabbits at 10 dpi (PI value of 79.2±18.0), and from three of four WNV-infected chickens at 14 dpi (PI value of 73.7±22.8). By contrast, sera collected between 0 and 4 dpi from WNV-infected rabbits were negative for WNV antibodies by NS1-cELISA (PI values of <40). From 14 to 21 dpi, three of the four infected chickens had antibody responses to WNV NS1 protein, whereas the remaining chicken was seronegative until the end of the study. Sera collected between 0 and 21 dpi from mock-infected rabbits and chickens were seronegative for WNV NS1. These results prove that the NS1-cELISA performs similarly to the in-house NT-ELISA test, as antibody responses to the E protein, but not NS1, were detected in animals inoculated with a killed virus (Fig. 2B and D).
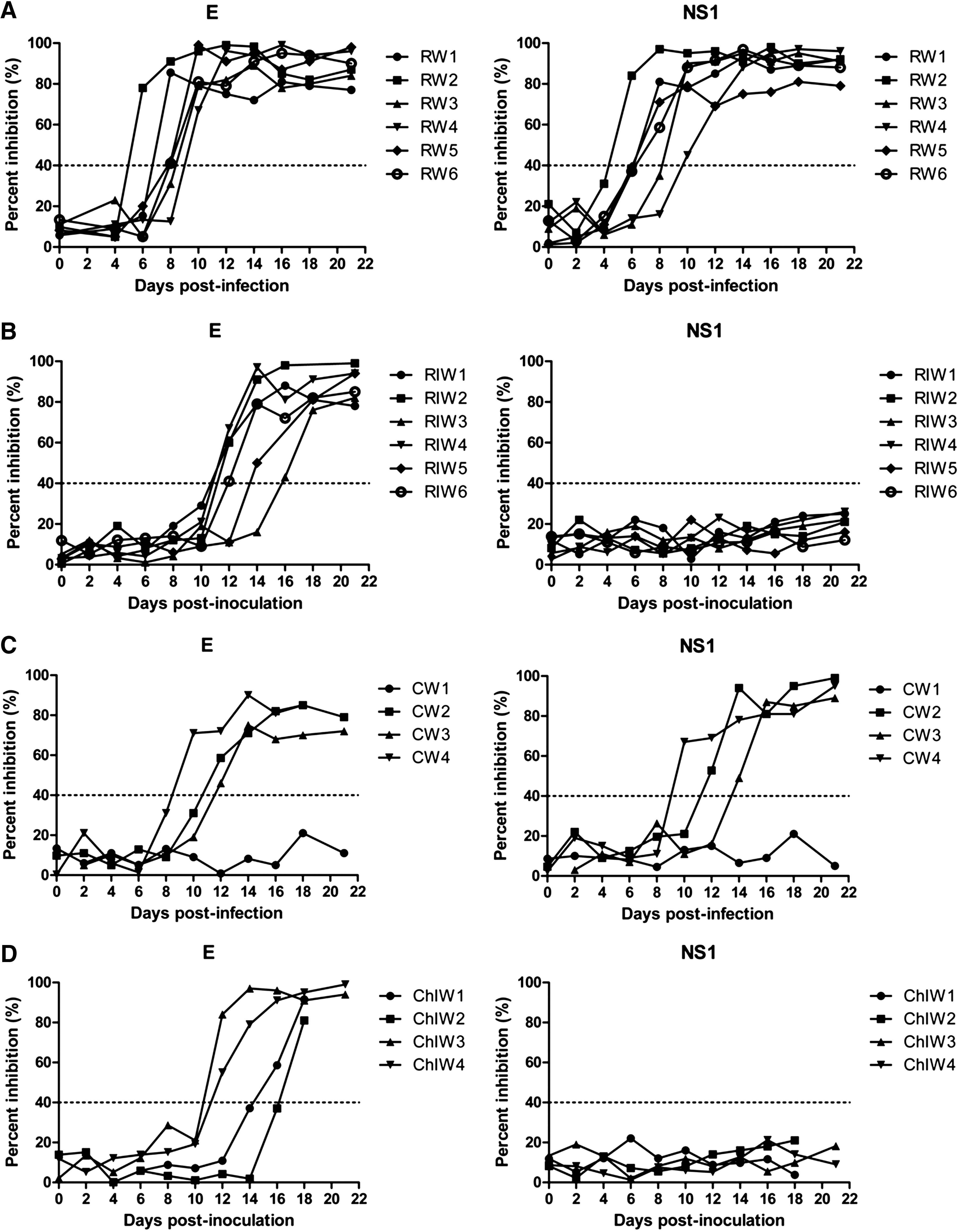
Kinetics of antibody development as determined by competitive enzyme-linked immunosorbent assay (NS1-cELISA), which detects antibodies against West Nile virus (WNV) non-structural protein 1 (NS1), and in-house IgG ELISA (NT-ELISA), which detects antibodies against WNV envelope (E) protein, in 12 rabbits and 8 chickens. Six rabbits and 4 chickens were infected with WNV (RW1 to RW6 and CW1 to CW4, respectively), and 6 rabbits and 4 chickens were vaccinated (RIW1 to RIW6 and ChIW1 to ChIW4, respectively). The cut-off percent inhibition (PI) value for rabbit sera was set at 40%.
Discussion
By using a bioengineered antigen that does not present a biohazard, the NS1-cELISA reduces the amount of manipulation of live WNV required for serology. In this study, the NS1-cELISA was able to clearly differentiate infected animals from vaccinated animals due to the presence of antibodies against NS1 induced by viral replication following infection. Therefore, the development of a test that differentiates between WNV infection and previous vaccination with a killed virus could greatly facilitate the diagnosis of early infections, and it could also enhance the use of WNV vaccines on a wider scale when WNV outbreaks occur in previously WNV-free regions.
Because the methods used to purify and concentrate antigens for vaccines can vary widely, whether traces of NSPs in vaccines may induce an anti-NSP antibody response remains controversial, as does whether they act as boosters in animals with low titers of antibodies to replicating viruses (Mackay et al. 1998). Some studies have reported that the inactivated virus vaccines that are currently available can induce the generation of NS1 antibodies in animals (Balasuriya et al. 2006; Kitai et al. 2010), whereas other studies have shown that the possibility of obtaining an anti-NSP antibody response from animals inoculated with an inactivated virus vaccine is very low (Mackay et al. 1998; Konishi et al. 2004). Regarding this difference, Japanese researchers commented that this phenomenon may be due to differences in preparation between the vaccines in terms of residual NS1 antigen (Kitai et al. 2010). In this study, the antibody response to NS1 was similar to that against envelope protein in WNV-infected rabbits and chickens, whereas animals inoculated with inactivated WNV developed antibody responses only to the envelope protein and not to NS1. We conclude that our competitive ELISA is adequately validated for the detection of anti-WNV NS1 antibodies in samples from various species. Our results show that the detection of antibodies against WNV NS1 will be useful for identifying viral activity in potential infections, and for evaluating the efficiency of control measures adopted during or after a disease outbreak.
WNV has an extremely broad vertebrate host range, including alligators (Klenk et al. 2004; Jacobson et al. 2005;), sheep (Kecskemeti et al. 2007), llamas (Kutzler et al. 2004), and alpacas (Dunkel et al. 2004; Kutzler et al. 2004), as well as some squirrel (Kiupel et al. 2003; Heinz-Taheny et al. 2004; Padgett et al. 2007; Gomez et al. 2008; Platt et al. 2008), chipmunk (Platt et al. 2007), and rabbit species (Tiawsirisup et al. 2005). Rabbits serve as dead-end hosts and have been found to develop viremia of sufficient magnitude to demonstrate even low competence for infection by feeding mosquitoes. Chickens are also susceptible to WNV infection and develop detectable antibodies (Senne et al. 2000; Langevin et al. 2001). Although the animals (rabbits and chickens) used in this study are known to be susceptible to WNV infection and to develop immune responses to it, the authors acknowledge that the horse is the best animal model. However, our findings provide a baseline for future investigations, and more studies in animal models including horses are required to gain a detailed understanding of the differential diagnosis of WNV. In addition, we found a monoclonal antibody (Mab b2NS22) lacking reactivity by IFA against lineage 1, but capable of reacting against lineage 2. This characteristic of this monoclonal antibody will require further investigation, and at present, experiments are underway to determine which factors are involved in this characteristic.
Footnotes
Acknowledgments
This work was supported by a grant from the National Veterinary Research and Quarantine Service, Ministry for Food, Agriculture, Forestry, and Fisheries of the Republic of Korea (grant number 6235-320-210-13).
Author Disclosure Statement
No competing financial interests exist.