
Abstract
Japanese encephalitis (JE) can infect many agriculturally important animals and humans, and has a high incidence in Asia. One of the natural hosts of the mosquito-borne JE virus (JEV) is domestic pigs, which act as amplifier hosts. Porcine infection results in fatal encephalitis, abortion, and stillbirth in pregnant sows, and hypospermia in boars. In this study, a rapid JEV detection method for swine and mosquitoes was developed based on reverse transcription loop-mediated isothermal amplification (RT-LAMP) targeting the nucleocapsid (E) genes of JEV genotype I (lineage K94PO5), and genotype III (lineage SA14-14-2). Fifty-six swine blood samples and 20,000 mosquitoes were used to evaluate the method, compared to conventional RT-polymerase chain reaction (PCR) and real-time RT-PCR. RT-LAMP had detection limits of 2.57 and 2.34 copies/μL for JEV I and III, respectively. Assay sensitivity was similar to real-time RT-PCR, but was 10-fold higher than conventional RT-PCR. Assay specificity was high, showing no cross-reactivity to other flaviviruses. The results of virus isolation and identification of swine blood samples and mosquito samples were fully consistent with RT-LAMP. Finally, the JEV RT-LAMP assay was simpler and less time consuming than conventional RT-PCR or real-time RT-PCR, since the amplification step could be completed in a single tube within 50 min at 63°C. In conclusion, the newly-developed RT-LAMP assay is an accurate and convenient method for rapid and sensitive diagnosis of JEV in swine and mosquitoes, and may prove to be a practical molecular tool for surveillance and epidemiological investigations.
Introduction
Diagnosis of JE by clinical symptoms is not feasible, due to non-specific signs at both the early and acute stages of infection. In addition, JEV isolation from peripheral blood is precluded by low-level transient viremia, which persists even in the acute stage of the disease (Shope and Meegan 1997). Currently, the most commonly used methods of swine diagnosis are laboratory-based enzyme-linked immunosorbent assays (ELISAs) that detect immunoglobulin (Ig)M or IgG antibodies in serum or cerebrospinal fluid taken from suspect herds (Yang et al. 2006). Unfortunately, these serological assays have considerable cross-reactivity with other flaviviruses (Holbrook et al. 2004).
Human JEV infection is clinically diagnosed by a variety of methods, including reverse transcription loop-mediated isothermal amplification (RT-LAMP). The RT-LAMP assay has been reported to be effective for detecting JEV in cerebrospinal fluid samples from patients with a clinical diagnosis of acute encephalitis (Parida et al. 2006). However, this approach has yet to be applied to a swine population. Genome-based JEV diagnostic assays have been previously developed for swine, and include the conventional RT-polymerase chain reaction (RT-PCR) and real-time RT-PCR. While both of these assays are specific and sufficiently sensitive, they require expensive thermal cycling equipment and technical expertise.
Therefore, we sought to develop a technically simple and highly accurate genome-based JEV diagnostic procedure for swine that is inexpensive and also easily applicable outside of the laboratory setting. To this end, the nucleocapsid (E) genes of JEV I (lineage K94PO5) and JEV III (lineage SA14-14-2) were selected as the detection targets in an RT-LAMP assay. The efficiency of this novel diagnostic procedure was comparatively analyzed with conventional RT-PCR and real-time RT-PCR for detecting JEV I and JEV III in swine and mosquitoes.
Materials and Methods
Viruses
JL0801 and YN0901 strains of JEV belong to the K94P05 and SA14-14-2 lineages, respectively, and they were selected as positive standards based on previous isolation from swine and mosquitoes.
Working stocks of the JL0801 and YN0901 strains were propagated in the mosquito cell line C6/36. Porcine stool specimens that contained porcine circovirus (PCV), porcine reproductive and respiratory virus (PRRSV), classical swine fever virus (CSFV), bovine viral diarrhea virus (BVDV), or swine hepatitis E virus (SHEV), were obtained from swine with laboratory-confirmed infections. A 10% (w/v) suspension of the respective stools were made and stored at −80°C until use. C. tritaeniorhynchus infected with Sindbis virus, chikungunya virus (CHIKV), or yellow fever virus (YFV) were confirmed and selected for study.
Sample collection
Fifty-six swine blood samples, including 46 positive sera and 10 negative serum samples, were obtained from different farms in the Li Shu County of Jilin Province (latitude 22°00’ N and longitude 100°47’ E), and were analyzed using molecular biological methods. C. tritaeniorhynchus adult female mosquitoes were trapped with ultraviolet lamps at dusk from cattle sheds and hog pens in the Jing Hong canton of Yun Nan Province (latitude 43°18’ N and longitude 124°20’ E). A total of 200 tubes of mosquitoes (100 mosquitoes/tube) were identified and sorted according to species, and only the C. tritaeniorhynchus females were used for these experiments.
Study methods
Gene amplification-based molecular techniques such as RT-PCR have emerged as a preferred method to make viral diagnoses using infected host tissue and fluid samples (Deubel et al. 1990; Tanaka 1993). However, since these methods require expensive equipment and technical expertise, they have not been adopted by many small and/or frontline laboratories. In addition, the low copy numbers of viral RNA in the blood of JEV-infected swine makes it difficult to extract sufficient amounts of RNA for such methods (Chuang et al. 2003).
RT-LAMP is a relatively new nucleic acid amplification method that can amplify DNA rapidly, efficiently, and with high specificity under isothermal conditions (Notomi et al. 2000). RT-LAMP relies on autocycling strand displacement DNA synthesis, which is carried out by Bst DNA polymerase large fragments with high strand displacement activity. The reaction is conducted under isothermal conditions that range from 60–65°C, and the amplified products can be readily analyzed by naked-eye visualization or digital imaging after electrophoresis through an agarose gel. In addition, detection can be carried out by evaluating the turbidity properties of the samples, caused by the large amounts of the by-product magnesium pyrophosphate from the amplification reaction, or by a colorimetric method that involves a visible color change produced by the addition of SYBR Green I to the amplification reaction (Mori et al. 2001; Nagamine et al. 2002).
Design of primers for RT-LAMP, real-time RT-PCR, and conventional RT-PCR, of JEV I and JEV III
For RT-LAMP amplification of the E gene sequences of JEV, four primers were designed based on alignment analysis of various JEV strains' genomic sequences (GenBank: Beijing-1, CJN-L1, JaOArS982, SA14–14–2, TL Taiwan, K94P05, and ThCMAr4492). Primer Explorer version 3 software was used (
JEV, Japanese encephalitis virus; RT-LAMP, reverse transcription loop-mediated isothermal amplification; RT-PCR, RT-polymerase chain reaction.
RNA extraction from the JL0801 and YN0901 strains, and reverse transcription to cDNA
The C6/36 cells were infected with the JL0801 and YN0901 strains. Culture supernatant containing the virus was harvested at 72 h post-infection. The supernatant was clarified of cellular debris by centrifugation. The RNA was extracted from 0.5 mL of culture supernatant containing 107 plaque-forming units of virus using the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany), in accordance with the manufacturer's protocol. In order to evaluate the detection limit of the assays, the amount of JEV RNA was determined by spectrophotometry and converted to molecular copies using a previously published formula (Krieg 1990):
Ten-fold serial dilutions of the JEV RNA (molecular copies from 106–10−1) were used as templates in reverse transcription reactions to synthesize cDNA. The 30-μL RT synthesis reaction for JEV consisted of 9.0 μL of DEPC H2O, 1.0 μL of 50 ng/μL genotype-specific reverse primer R1 or R2, and 5.0 μL of total RNA. The mixture was incubated at 75°C for 5 min, then chilled on ice for at least 5 min. Afterwards, the following reagents were added to each of the reactions: 9.0 μL of DEPC H2O, 1.0 μL of 200 U/μL M-MLV RT (Promega, Madison, WI), 1.0 μL of 40 U/μL ribonuclease inhibitor (Promega), 2.0 μL of 5×RT buffer, and 2 μL of 10 mM dNTP mix. The reaction mixture was incubated at 42°C for 50 min and 75°C for 5 min.
The resultant cDNA products were concentration detected by a NanoDrop 2000 spectrophotometer (Thermo-Scientific, Kalamazoo, MI), and used as a template in RT-LAMP reactions.
RT-LAMP reaction and product detection
RT-LAMP was carried out in a 25-μL reaction mixture that contained 1.0 μL of 5 μM each of the F3 and B3 primers, 1.0 μL of 40 μM each of the BIP and FIP primers, 2.5 μL of 10×ThermoPol reaction buffer, 1.0 μL of 8 U/μL Bst DNA polymerase (New England Biolabs, Ipswich, MA), 2.5 μL of 10 mM dNTP mix (TaKaRa, Shiga, Japan), 4.0 μL of 5 M Betaine (Sigma-Aldrich, St. Louis, MO), 5.0 μL of 30 mM MgSO4, (Invitrogen, Carlsbad, CA), 4.0 μL of target cDNA, and 2 μL of nuclease-free water. The amplification reaction was performed at 65°C for 60 min, and terminated by heating to 80°C for 10 min. Sterile water was used as a negative control template. RT-LAMP products were resolved by 2.0% agarose gel electrophoresis, and visualized by ethidium bromide staining and ultraviolet (UV) light detection; the positive reaction mixtures showed a characteristic ladder-like pattern. In an alternative protocol, the products were observed directly with the naked eye by adding 1.0 μL of SYBR Green I (Invitrogen) to the reaction mix. The solution turned green in the presence of RT-LAMP amplification products, but stayed orange in the absence of the amplicon.
Optimization of the RT-LAMP assay
The RT-LAMP reaction mixtures were incubated at 60, 61, 62, 63, 64, or 65°C for 60 min to determine the optimal reaction temperature. Subsequently, the optimal reaction time was determined by performing RT-LAMP at 63°C for 30, 35, 40, 45, 50, or 60 min. The reactions were terminated by heat inactivation at 80°C for 10 min. The amplified RNA products from the RT-LAMP assays were visualized, as described above, by agarose gel electrophoresis or SYBR Green I.
Determination of the RT-LAMP assay detection limit
To determine the sensitivity of the RT-LAMP assay, the 10-fold serial dilutions of the JEV I and JEV III RNA molecular copies (from 106–10−1) were reacted. The various reaction mixtures were incubated at 63°C for 50 min, followed by termination at 80°C for 10 min. RT-LAMP products were analyzed by the electrophoresis and SYBR Green I detection methods.
Determination of the real-time PCR assay detection limit
Real-time PCR was performed with the JEV I- and JEV III-specific primers (JEVYG1/YG2 and JEVYG3/YG4, respectively). Amplification was carried out in a 25-μL total reaction volume consisting of 12.5 μL of SYBR Green Real-Time PCR Master Mix (Toyobo, Osaka, Japan), 8.0 μL of nuclease-free water, 1.0 μL of 0.4 μM forward and reverse primers, and 5 μL of the cDNA sample (molecular copies from 106–10−1). The following real-time RT-PCR thermal cycling program was run on an ABI Prism 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA): one cycle of 50°C for 2 min and 95°C for 5 min, followed by 40 cycles of 95°C for 15 sec, 60°C for 15 sec, and 72°C for 50 sec. The data were analyzed with the accompanying ABI 7500 software package, version 2.0.5.
Determination of the PCR assay detection limit
PCR was performed with the JEV I- and JEV III-specific primers that targeted the respective E genes. The reaction was carried out in a 20-μL total reaction volume that contained 1.0 μL of 5 U/μL ExTaq polymerase (TaKaRa), 0.5 μL of 10 mM dNTP mix, 0.5 μL each of 10 μM primers JEV E1 and E2 or JEV E3 and E4, 2.0 μL ExTaq buffer, 4.0 μL cDNA, and 11.5 μL nuclease-free water. The PCR reaction was carried out on a TC-512 instrument (Techne, Staffordshire, U.K.) using the following thermal cycling conditions: one cycle of 94°C for 5 min, followed by 30 cycles of 94°C for 30 sec, 56°C for 30 sec, and 72°C for 30 sec, and a final extension cycle of 72°C for 10 min. The products were analyzed by 1.0% agarose gel electrophoresis with ethidium bromide staining and UV light-assisted visualization. The expected amplicon sizes were 475 bp (JEV I) and 524 bp (JEV III).
Specificity of the RT-LAMP assay
To analyze the specificity of the established RT-LAMP assay, JEV I (JL0801) and JEV III (Yunnan 0901), PCV (NM), PRRSV (CC-1), CSFV (PR-2008), BVDV (HLJ-10), SHEV (CHN-XW), CHIKV (YN1011), YFV (YN1001), and Sindbis virus (YN87488), were used as templates and subjected to RT-LAMP as described above. The reaction was performed at 63°C for 50 min.
Virus isolation and identification of swine blood samples and mosquito samples
Emulsions [10% (w/v)] of suspensions of 56 swine blood samples, including 46 positive samples and 10 negative serum samples, and 200 tubes of mosquito suspensions were prepared in Eagle's minimum essential medium (EMEM) that contained 2% heat-inactivated fetal bovine serum (FBS). The isolate was propagated in Aedes albopictus C6/36 cells. The cytopathogenic effects (CPEs) were observed in the C6/36 cell cultures inoculated with the emulsion made from the mosquito and swine blood suspensions. Viral RNA was reverse transcribed and PCR-amplified with PPM1 and PPM2 primers using the OneStep RT-PCR kit (Qiagen). These primers amplify the partial capsid/premembrane (C/prM) gene of JEV (Chung et al. 1996). RT-PCR was performed according to the manufacturer's instructions and the method described in a previous report (Chung et al. 1996). RT-PCR products with the expected size (198 base pairs) corresponding to the partial C/prM gene of JEV from all examined samples were identified in 2.0% agarose gel.
Results
RT-LAMP amplification of the E gene from JEV I and JEV III and assay specificity
JEV RNA and the genotype-specific primers that targeted the virus E gene from the JL0801 and YN0901 strains were included in an RT-LAMP assay that was performed in a 65°C water bath for 60 min. The agarose gel electrophoresis analysis indicated that the amplified DNA products produced a characteristic laddering pattern with multiple bands, which indicated that the final RT-LAMP products were mixtures of stem-loop DNAs with various stem lengths (Fig. 1A and B). In contrast, the negative control did not produce the characteristic multi-band laddering pattern.
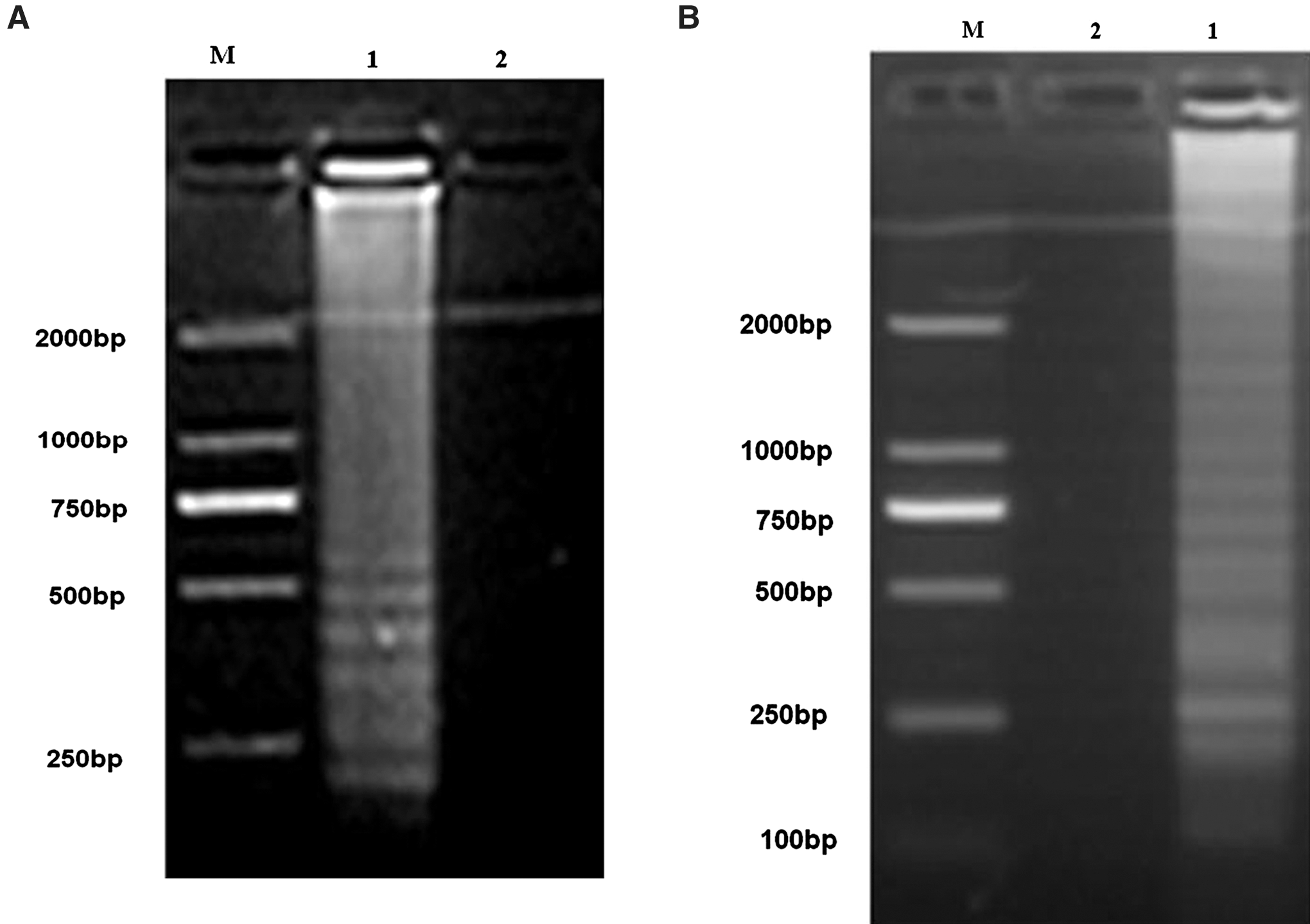
Amplification of the E gene from JEV I (
Optimization of the RT-LAMP assay
The optimal reaction temperature and time of the RT-LAMP assay were investigated. As shown in Figure 2A and B, the products of the RT-LAMP assay at different temperatures produced the characteristic multi-band laddering pattern. However, the intensity of bands in the ladder was the strongest from the reactions carried out at 63°C, indicating that this is the optimal temperature for RT-LAMP amplification of JEV E genes from JL0801 and YN0901.

Determination of optimal conditions for RT-LAMP detection of JEV I (
The RT-LAMP assay was then performed at the 63°C optimal temperature for different lengths of time. The subsequent results indicated that the DNA products with the highest intensity were produced using a 50-min reaction time (Fig. 2C and D). Therefore the optimal reaction conditions of the RT-LAMP assay for detecting JEV was determined to be 63°C for 50 min.
Detection limit of the RT-LAMP assay compared with conventional RT-PCR
The sensitivity of the E-specific RT-LAMP assay was then compared with that of conventional PCR using E-specific primers. The JEV I and JEV III RNA initial concentrations were converted to 2.57×106 copies/μL and 2.34×106 copies/μL, respectively. Then 10-fold serial dilutions of the JEV I and JEV III RNA molecular copies (from 106–10−1) were used to determine assay sensitivity.
As observed by 2% agarose gel electrophoresis, the JEV I and III detection limits of the RT-LAMP assay were estimated to be 2.57 copies/μL and 2.34 copies/μL, respectively (Fig. 3A and B). The RT-LAMP assay was more sensitive than the conventional RT-PCR assay (Fig. 3C and D). In addition, visual inspection of amplification products using SYBR Green I stain was better for the RT-LAMP assay than the conventional PCR assay (RT-LAMP detection sensitivity: 2.57 JEV I copies/μL and 2.34 JEV III copies/μL versus conventional RT-PCR; both p<0.05; Fig. 3E and G). However, the SYBR Green I staining method was inferior to the electrophoresis detection method. Using UV transillumination allowed RT-LAMP detection limits of 2.57 JEV I copies/μL and 2.34 JEV III copies/μL (Fig. 3F and H; Table 2).
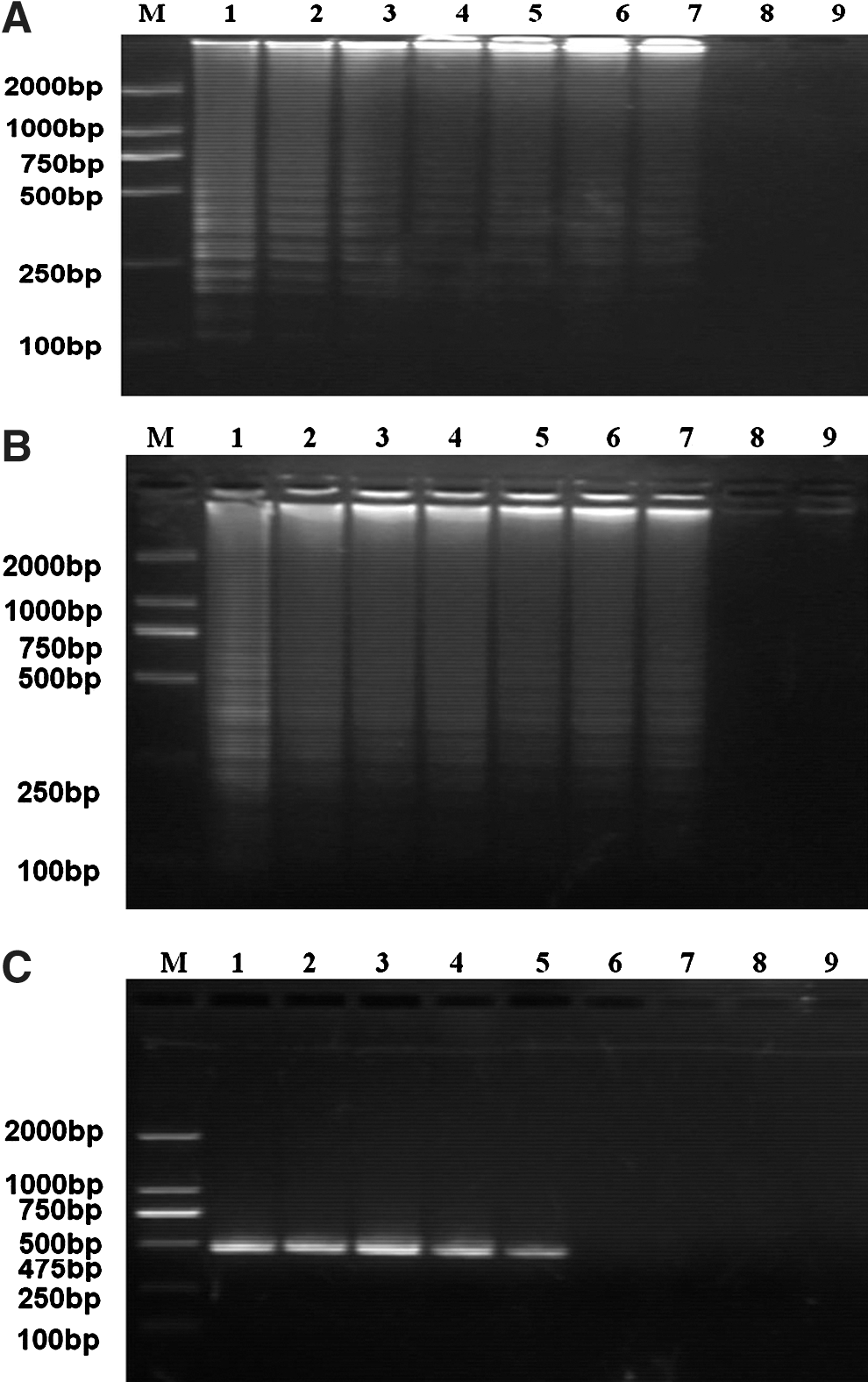
Sensitivity of the RT-LAMP assay for the JEV I and JEV III E genes, as detected by electrophoresis and SYBR Green I staining. Shown are the RT-LAMP 2% agarose gel electrophoresis detection limits for JEV I (
+, positive reaction; −, negative reaction; JEV, Japanese encephalitis virus; RT-LAMP, reverse transcription loop-mediated isothermal amplification; RT-PCR, RT-polymerase chain reaction.
Detection limit of the RT-LAMP assay compared with real-time RT-PCR
Serial 10-fold dilutions of JEV I and JEV III RNA were tested in parallel by real-time RT-PCR. The detection limits of conventional real-time RT-PCR were 2.57 copies/μL and 2.34 copies/μL, respectively (Fig. 4A and B). Figure 5 shows the standard curves for JEV I (Fig. 5A) and JEV III (Fig. 5B), which were constructed using the known concentrations of the respective 10-fold serial dilutions. Melting curve analysis showed that specific amplification melting temperatures were 87.3°C (for JEV I; Fig. 6A) and 86.6°C (for JEV III, Fig. 6B). According to the amplification and dissociation plots, the amplified products could be identified as specific genomic sequences. Compared to products from the RT-LAMP assay, the detection sensitivities by agarose gel electrophoresis and SYBR Green I staining were equivalent with those of real-time RT-PCR. The details are summarized in Table 3. Moreover, the RT-LAMP amplification procedure was completed within 50 min, which was much shorter than the 2.5 h required for the real-time RT-PCR procedure.

Detection sensitivity of the real-time RT-PCR assay for JEV I (
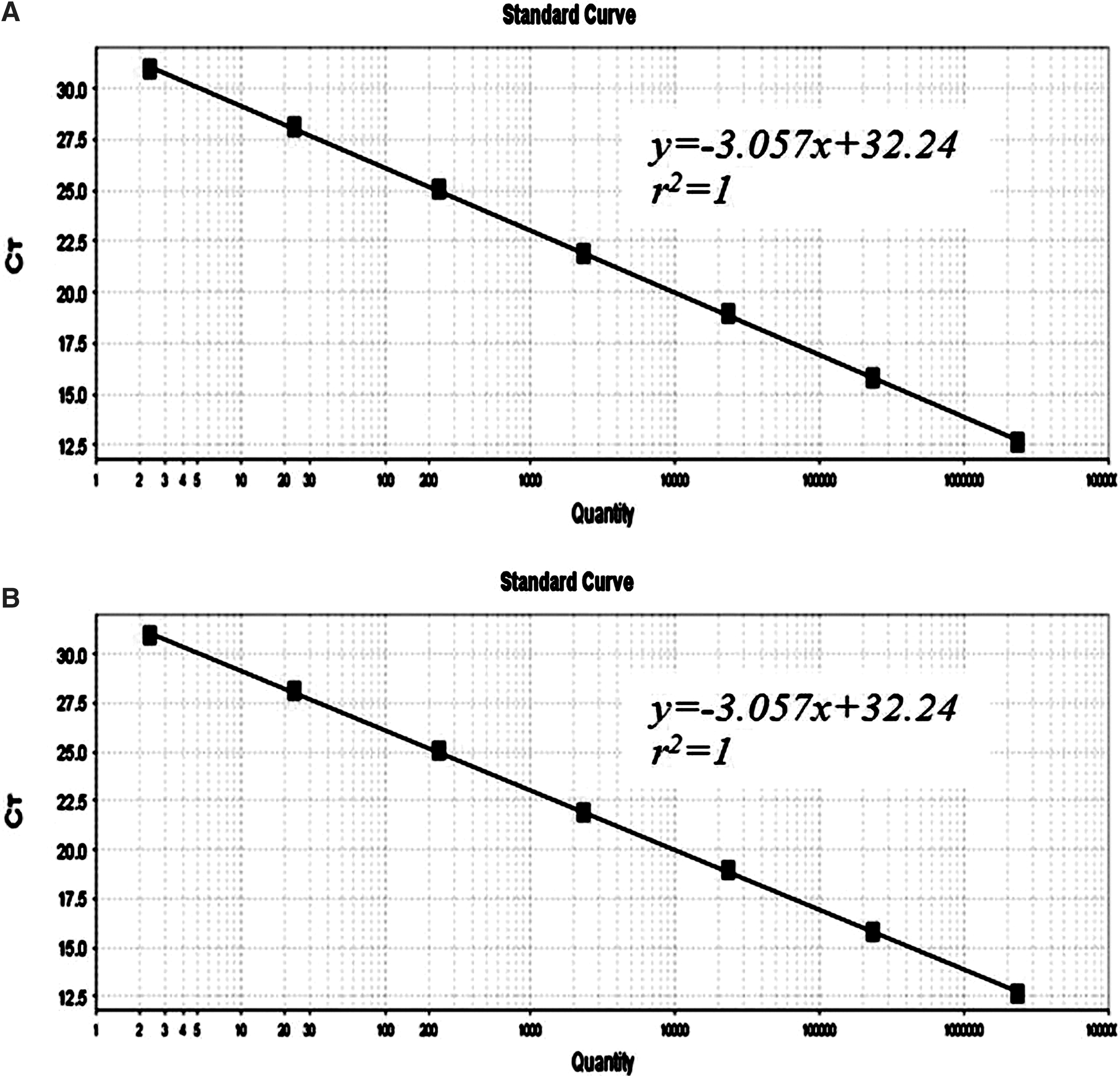
Standard curve of real-time RT-PCR for detecting JEV I (
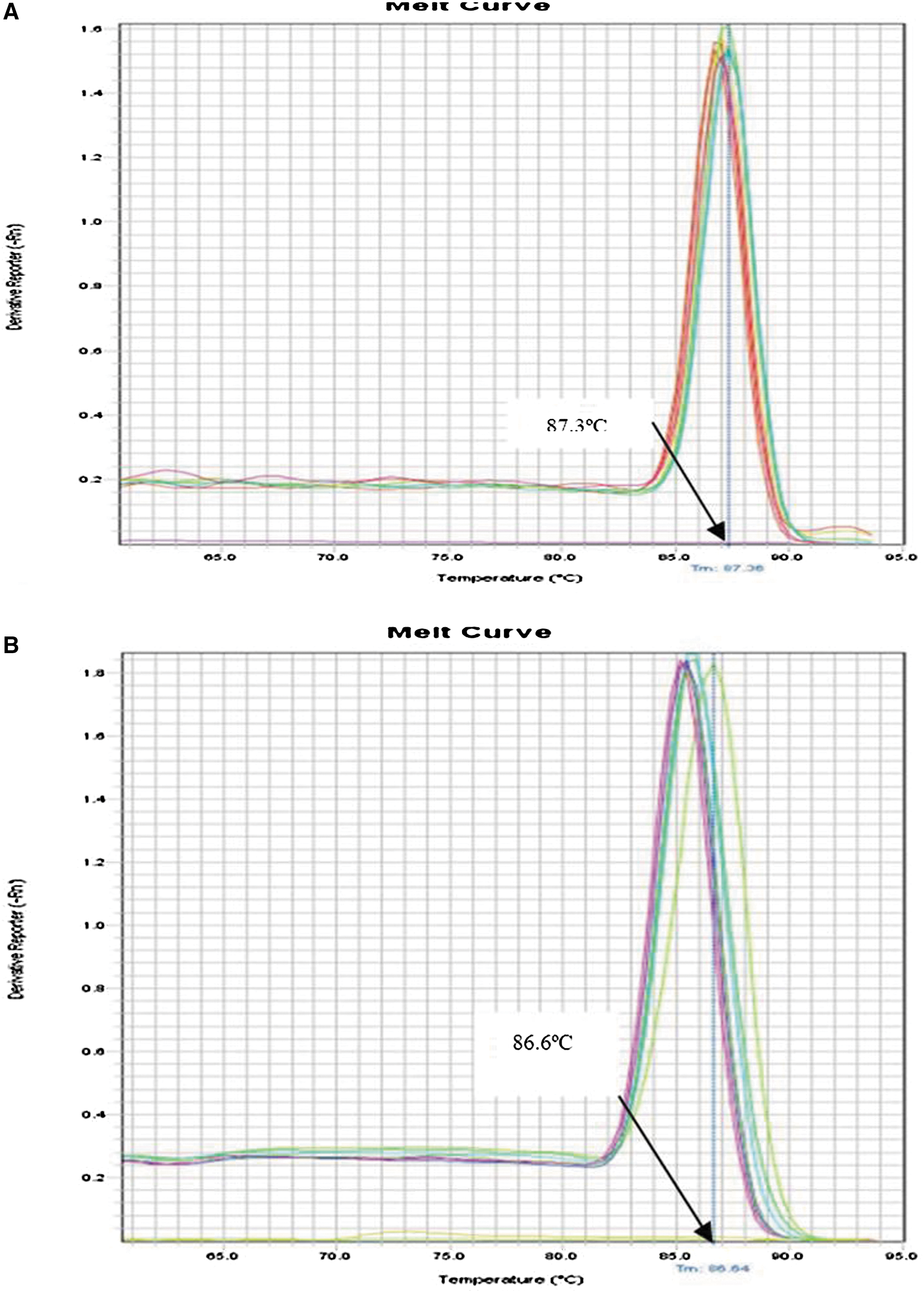
Dissociation curve of real-time RT-PCR for detecting JEV I (
+, positive reaction; −, negative reaction; JEV, Japanese encephalitis virus; RT-LAMP, reverse transcription loop-mediated isothermal amplification; RT-PCR, RT-polymerase chain reaction.
Specificity of the RT-LAMP assay
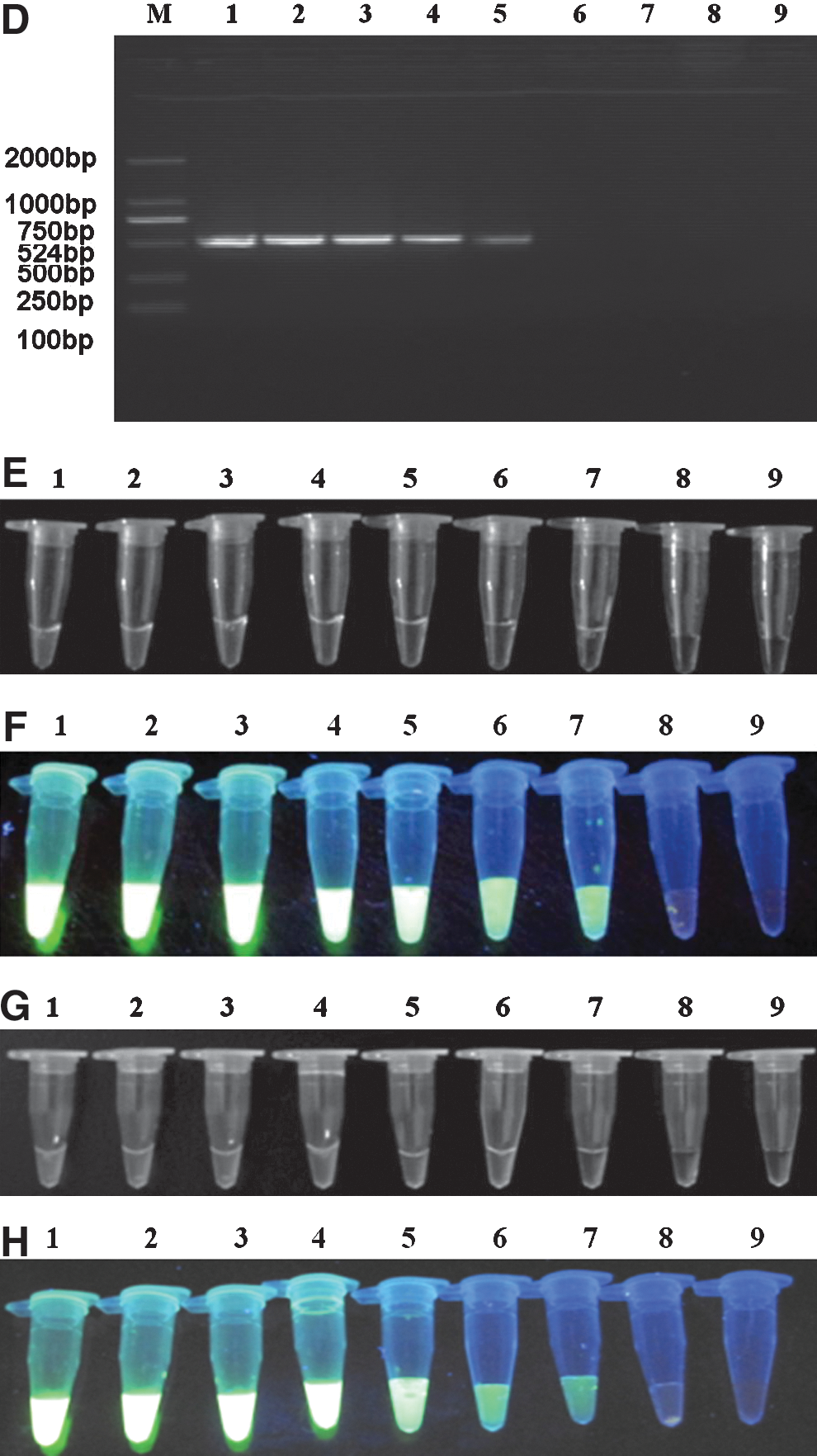
Figure 7 shows that the RT-LAMP assay with JEV genotype-specific primers produced no positive reactions when using PCV, PRRSV, CSFV, BVDV, SHEV, CHIKV, YFV, and Sindbis virus as templates.

Specificity of the RT-LAMP assay for detecting the JEV I and JEV III E genes. JEV and other selected viruses were used as templates and subjected to RT-LAMP performed at 65°C for 60 min (lanes: M, DL2000 DNA marker; 1–10, RT-LAMP results from templates JEV I, JEV III, PCV, PRRSV, CSFV, BVDV, SHEV, CHIKV, YFV, and Sindbis virus, respectively; 11, negative control).
Validation of the JEV RT-LAMP assay with swine blood samples and mosquito samples
We tested the field applicability of the RT-LAMP assay by using positive sera and mosquitoes taken from local agricultural hog pens with ongoing JEV epidemics. The results were compared with those from conventional RT-PCR and real-time RT-PCR. The samples (true-positive or false-positive) were determined by the results of isolation and identification of JEV.
The LAMP assay demonstrated much higher sensitivity than conventional RT-PCR, but had similar sensitivity to real-time RT-PCR (Tables 4 and 5). All 10 negative serum samples were also negative by all of the tests, thereby ruling out the possibility of false-positives. None of the RT-PCR- or real-time RT-PCR-positive samples was missed by RT-LAMP, thereby indicating the higher sensitivity of the RT-LAMP assay. Results of virus isolation and identification of swine blood samples and mosquito samples were consistent with RT-LAMP results. The results of swine blood samples by RT-PCR and real-time RT-PCR were confirmed to be 2 false and 1 false for JEV I-negative, and 6 false and 1 false for JEV III-negative, respectively. The results of mosquito samples by RT-PCR and real-time RT-PCR were confirmed to be 2 false and 0 false for JEV I-negative, and 14 false and 4 false for JEV III-negative, respectively.
JEV, Japanese encephalitis virus; RT-LAMP, reverse transcription loop-mediated isothermal amplification; RT-PCR, RT-polymerase chain reaction.
JEV, Japanese encephalitis virus; RT-LAMP, reverse transcription loop-mediated isothermal amplification; RT-PCR, RT-polymerase chain reaction.
Discussion
JEV is an important pathogen of humans and agricultural animals. JEV infection in pigs causes severe reproductive disorders in both male and female pigs. Accurate diagnosis of JE in suspected encephalitis infections is essential for optimal disease management and protection of the remaining herd. However, the application of conventional assays, such as RT-PCR, real-time RT-PCR, oligonucleotide microarray, and ELISA, has been limited by their requirements for laboratory-based operations, skilled technicians, and expensive equipment. A high-throughput method that can be readily adapted for use on pig farms, especially in remote rural areas where there is a high risk of JEV infection, is urgently needed for effective surveillance of new circulating JEV strains.
The RT-LAMP assay has the advantages of reaction simplicity and detection sensitivity, as the reaction is carried out in a single tube by isothermal incubation; the optimal reaction parameters for JEV E gene detection were 63°C and 50 min. In contrast to conventional RT-PCR, real-time RT-PCR, oligonucleotide microarray, and ELISA, the RT-LAMP method does not rely on expensive or sophisticated facilities or equipment, such as thermal cyclers. The amplification products of the reaction produced characteristic ladder-like patterns by agarose gel electrophoresis, or were detectable by naked-eye inspection with SYBR Green I under normal light or UV transillumination.
Herein, we describe the establishment of a novel two-step RT-LAMP assay that can rapidly detect the E genes of JEV I and JEV III in swine and mosquitoes, and has higher sensitivity than conventional RT-PCR and real-time RT-PCR. In addition, the JEV RT-LAMP assay showed good specificity, as no positive results were found in the negative control samples, which included PCV, PRRSV, CSFV, BVDV, SHEV, CHIKV, YFV, and Sindbis virus assayed under the same experimental conditions. However, since only Chinese JEV strains were tested, more research is required to determine how the RT-LAMP assay performs with all known strains of JEV.
In summary, we describe a LAMP method for the detection of JEV I and JEV III in swine that is rapid, sensitive, and specific. From a practical point of view, this method is more suitable than the currently available methods for use as a routine diagnostic tool, and can accommodate the large numbers of clinical samples required by active pig farms.
Footnotes
Acknowledgments
We wish to express our sincere gratitude to the Yunnan Institute of Parasitic Diseases for assistance in mosquito collection, and especially to Hong-Ning Zhou and Zhong-Hua Yang for their excellent technical assistance. This work was supported by a grant from the Special Fund for Agro-Scientific Research in the Public Interest (no. 201203082).
Author Disclosure Statement
No competing financial interests exist.