
Abstract
Genotyping of bacteria is critical for diagnosis, treatment, and epidemiological surveillance. Coxiella burnetii, the etiological agent of Q fever, has been recognized to have a potential for bioterrorism purposes. Because few serosurveys have been conducted in Italy, there is still limited information about the distribution of this pathogen in natural conditions. In this paper, we describe the genotyping of C. burnetii strains by multispacer sequence typing (MST) detected in cattle and goat farms in the Abruzzi region of Italy. Biological samples (milk, aborted fetus) positive for C. burnetii DNA were sequenced in the spacer regions and compared with those already publicly available (
Introduction
C
oxiella burnetii,
Humans are also susceptible to Q fever. The main sources of infection for humans are feces, urine, and products of conception of infected animals. The bacteria may persist in the environment for years (Raoult et al. 2005, Tissot-Dupont and Raoult 2008). Moreover, the amniotic fluid and placenta of infected animals contain high titers of bacteria that may spread in the environment through aerosol particles remaining virulent for months (Sting et al. 2013). Infection in humans can also be acquired via ingestion of unpasteurized milk or other contaminated material (Masala et al. 2004), although controversy remains concerning the possibility of human infection by the oral route (Arricau-Bouvery and Rodolakis 2005, Angelakis and Raoult 2010, EFSA 2010).
To prevent disease in humans, it is critical to trace the source of each Q fever outbreak. For this reason, several genotyping methods have been described thus far. Until 2005, these techniques were based on plasmid typing (Willems et al. 1993, Valkovà and Kazàr 1995, Jäger et al. 2002), restriction fragment length polymorphism and pulsed-field gel electrophoresis (RFLP-PFGE) analysis (Vodkin et al. 1986, Hendrix et al. 1991, Jäger et al. 1998), and sequence analysis of individual genes such as16S, 23S (Stein et al. 1997), com1, mucZ, and icd (Zhang et al. 1997, Nguyen and Hirai 1999, Sekeyovà et al. 1999). Nevertheless, some of these methods exhibited limitations on inter- and intralaboratory reproducibility that hindered their widespread use (Massung et al. 2012). In 2005, typing via sequence analysis of the multispacer region (multispacer sequence typing [MST]) was described (Glazunova et al. 2005).
The aim of this study was to quantify the presence of the infectious agent by qPCR and to genotype by MST C. burnetii strains detected in three unrelated farms in the Abruzzo region of central Italy. The results suggest circulation of multiple strains and provide further insights into C. burnetii epidemiology and evolution.
Materials and Methods
Biological samples and DNA extraction
Between October 10, 2012 and February 26, 2013, 20 milk samples (n=20) of cattle were collected in a farm in Pescara Province (farm 1) in which metritis and retained fetal membranes were noticed in several animals by the local veterinarian. In 2012, seven milk samples (n=7) were collected in a goat farm in Scanno, L'Aquila Province (farm 2). The goats showed symptoms consistent with the APSW complex. The temperature of samples was maintained between 4°C and 8°C until tested. Once transferred into 50-mL tubes, the milk was centrifuged for 10 min at 3000×g. The supernatant was discarded, and 300 μL of the semisolid pellet was distributed into the cartridge provided by the kit for the automated DNA extraction (Maxwell® 16 Cell DNA Purification Kit, Promega). A third farm, located in Avezzano, L'Aquila province (farm 3), was investigated in 2013. Brain, spleen, and lung of an aborted goat fetus were collected and used for DNA extraction by means of the Maxwell 16 Tissue DNA Purification Kit (Promega). DNA aliquots were stored at −20°C until use. Farms are located in Abruzzo region, Central Italy. The geographical distribution is illustrated in Figure 1.
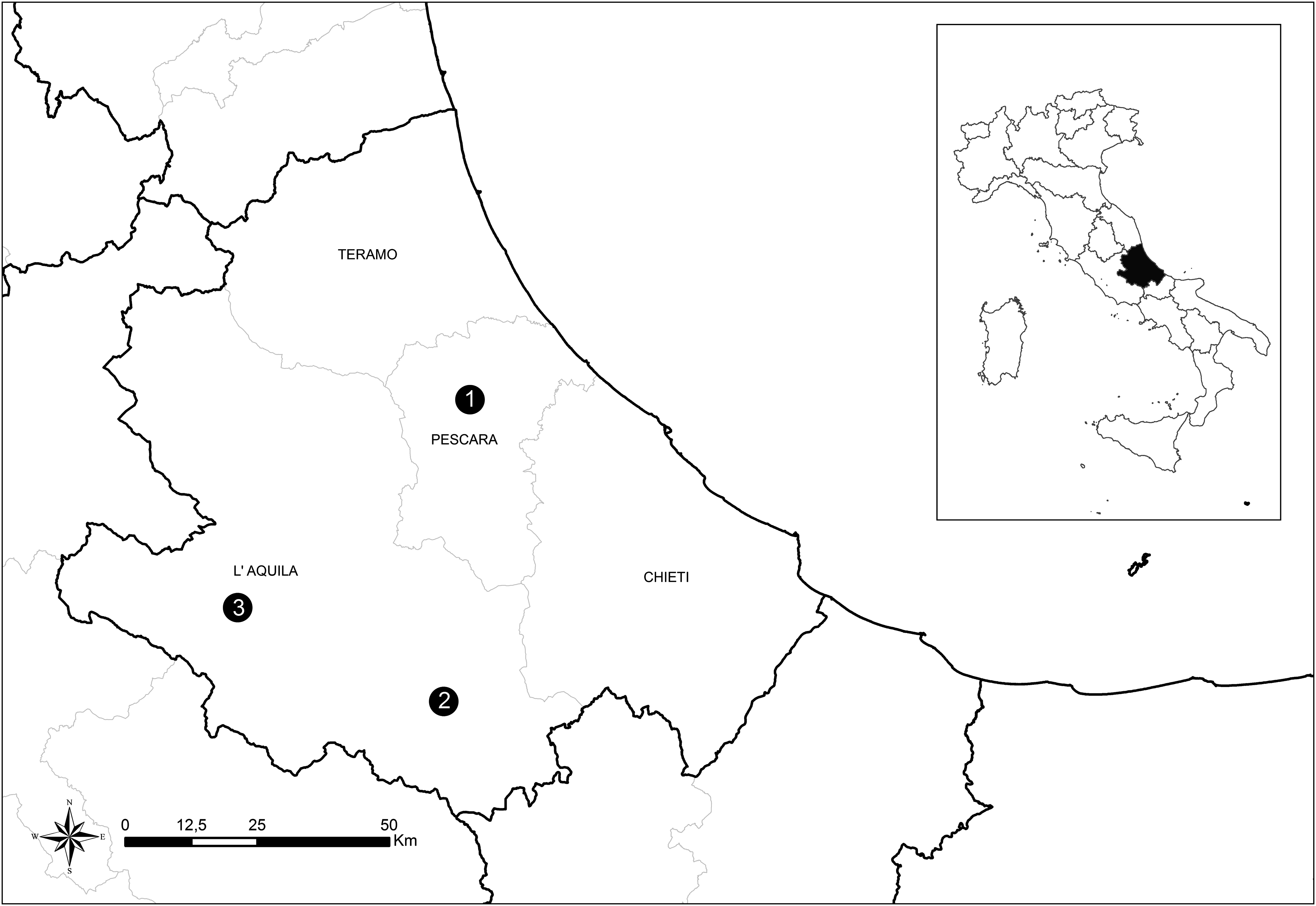
Map of the sampling area. Geographical distribution of the three farms in the Abruzzo region, Central Italy. (1) Cow farm 1 located in Pescara province; (2) goat farm 2 located in Scanno, L'Aquila province; (3) goat farm 3 located in Avezzano, L'Aquila province.
Detection of C. burnetii by real-time PCR
All samples were tested by real-time PCR to detect the presence of C. burnetii as previously described (Panning et al. 2008) with some modifications. In particular, the probe was modified as follows: FAM-TGCATAATTCATCAAGGCACCAATGGT-TAMRA (Eurofins Mwg Operon, Inc.). The 20-μL reaction mixture contained 2×TaqMan Fast Universal PCR Master Mix, 300 nM probe, 900 nM primers, nuclease-free water, and 5 μL of DNA. Real-time PCR was performed using the 7900HT Fast Real Time PCR System (Applied Biosystems). The amplification protocol used was: 20 s at 95°C followed by 35 cycles of 1 s at 95°C and 20 s at 60°C.
Evaluation of C. burnetii concentration in milk by quantitative real-time PCR
Milk samples were analyzed by quantitative real-time PCR (qPCR) to assess the level of shedding of C. burnetii in infected animals. The qPCR was performed by using the LSI VetMAX™ Coxiella burnetii Real-Time PCR Kit, absolute quantification (Life Technologies, USA) following the manufacturer's instructions. Lactating cows showing excretion levels ++ and +++ were considered heavy shedders according to Guatteo et al. (2007).
Multispacer sequence typing and phylogenetic analysis
Molecular characterization of C. burnetii DNA was performed as previously described (Glazunova et al. 2005) with some modifications. Milk samples (n=2) from cattle (nos. 32 and 303 collected in farm 1), milk (n=1) from goat (collected from farm 2), and the brain of the aborted fetus (farm 3), that were shown to possess the highest DNA concentration, were selected for MST assay. Each PCR was carried out in a GeneAmp PCR System 9700 (Applied Biosystems). Five microliters of the extracted DNA were amplified in a 50-μL reaction mixture containing 200 nM of each primer (Glazunova et al. 2005), 200 μM deoxynucleotide triphosphates (dNTPs; Promega), 2.5 mM MgCl2 Solution (Applied Biosystems), 0.03 U/μL AmpliTaq Gold™ (Applied Biosystems), and 1× PCR Buffer II (Applied Biosystems). Amplifications were carried out under the following conditions: Initial denaturation of 10 min at 95°C, followed by 40 cycles of denaturation for 30 s at 95°C, annealing for 30 s at 57°C, and extension for 30 s at 72°C. The final extension was performed at 72°C for 7 min.
The reaction mix for the Cox57 spacer was modified by designing a novel forward primer (5′-CCTGGACCGAGAGCACAAAC-3′), using a final concentration of 600 nM for both forward and reverse primers, and setting the annealing temperature at 60°C. For the Cox56 spacer, only the primer concentration was modified (600 nM each). PCR products were purified using the QIAquick PCR Purification Kit (Qiagen) and sequenced by BigDye Terminator v.3.1 (Applied Biosystems) and the 3130 XL Genetic Analyzer (Applied Biosystems). Raw sequence data were assembled using Contig Express (Vector NTI suite 9.1; Invitrogen). The coded alleles were compared with the sequences in the reference database available on the website
The sequences publicly available from the 34 groups listed in the MST database, and the sequences coming straight from DNA samples of the present study were employed for phylogenetic analysis. Sequence alignment for assembled spacer sequences and phylogenetic analyses were conducted using MEGA version 6 (Tamura at al. 2013), and the evolutionary distances were computed using the Tajima–Nei method (Tajima and Nei 1984).
Results
Detection of C. burnetii by real-time PCR
Fifteen of 20 milk samples of cattle, four of seven milk samples of goat, and the gross organs of the fetus were positive when tested for the presence of C. burnetii by real-time PCR (Table 1).
Fifteen cattle milk and four goat milk samples positive for IS111 were analyzed to assess the level of excretion of C. bunetii in infected milk. Cycle threshold (Ct) values correspond to the average of three replicates.
Excretion levels according to the LSI VetMAX™ Coxiella burnetii Real-Time PCR Kit, absolute quantification (Life Technologies, USA): +, weak excretion; ++, high excretion; +++, very high excretion;
Ct, cycle threshold; NQ, not quantifiable (Ct values below the limit of quantification).
Evaluation of C. burnetii concentration in milk by qPCR
Milk samples that turned out to be positive were also tested by qPCR to quantify Coxiella DNA. Three milk samples showed a signal below the limit of quantification, whereas the remaining samples showed a bacterial concentration ranging from 102 bacteria/mL to 105 bacteria/mL. Milk samples from cattle 32 and 303 (farm 1) showed estimated titers of C. burnetii above 105 bacteria/mL that were not correctly quantifiable because they were out of the quantification range of the standard curve (Table 1).
MST and phylogenetic analysis
Coxiella DNA isolated from milk samples of cattle numbers 32 and 303 (farm 1) confirmed 100% sequence similarity within them. The MST profile is similar to that of sequence type (ST) 20 (Glazunova et al. 2005) (Fig. 2), with the exception of the Cox57 spacer sequence in which a mutation T/A at position 549 of allele 6 is present. These data suggest the presence of a new allele, implying the existence of a new ST. The sequence of the new allele for Cox57 was deposited in GenBank (accession no. KF781199).
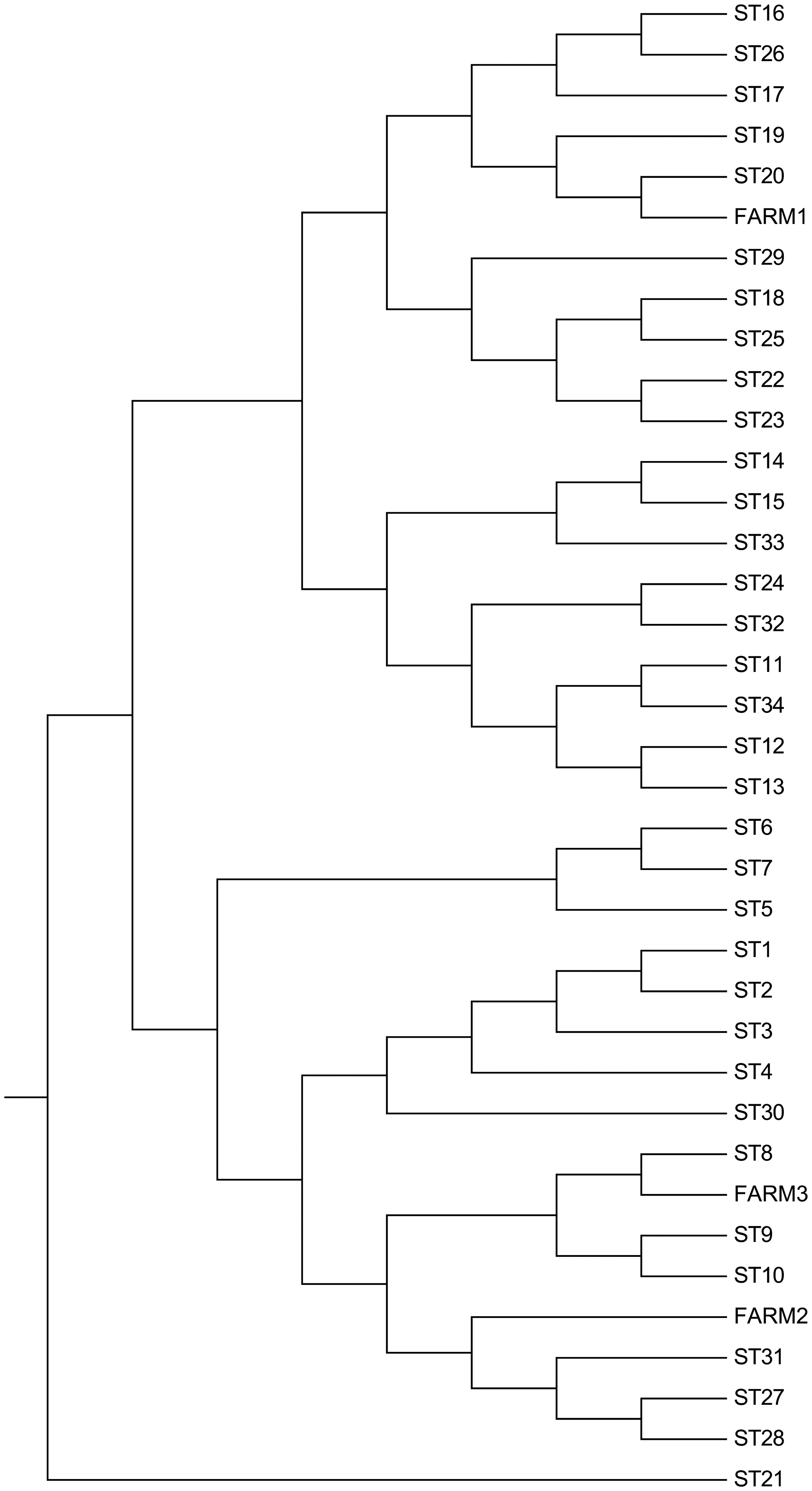
Phylogenetic analysis. The evolutionary history was inferred using the unweighted pair group method with arithmetic mean (UPGMA) method (Sneath and Sokal 1973). The optimal tree with the sum of branch length=0.02463836 is shown. The evolutionary distances were computed using the Tajima–Nei method (Tajima and Nei 1984) and are in the units of the number of base substitutions per site. The analysis involved 37 nucleotide sequences. All positions with less than 0% site coverage were eliminated; that is, fewer than 100% alignment gaps, missing data, and ambiguous bases were allowed at any position. Evolutionary analyses were conducted in MEGA6 (Tamura at al. 2013). The tree was drawn according to Hornsta et al. (2011) and rooted according to Pearson et al. (2013).
Coxiella DNA isolated from the goat milk sample number 3 from farm 2 has a new ST because it showed a novel combination of alleles. This novel MST group forms a monophyletic group within a wider cluster, as depicted in the phylogenetic tree. Indeed, its profile differs significantly from the existing strains publicly available (Fig. 2). Coxiella spacer sequences from the fetus brain (farm 3) did not show new alleles. However, the allelic combination elicited a novel ST profile very similar to ST8 (Fig. 2). A summary of the novel ST profiles detected in this study is shown in Table 2.
The genotype of C. burnetii from cattle milk samples (farm 1) was similar to sequence type 20 (ST20), with the exception of the Cox57 spacer sequence in which a novel allele was described in the present study. Different alleles were not found in C. burnetii from goat fetus (farm 3) and goat milk (farm 2); however, in both cases the allelic combination generated unique STs.
Discussion
In this study, we described the detection of C. burnetii by real-time PCR in samples collected from animals of three unrelated farms of central Italy showing symptoms consistent with C. burnetii infection. Furthermore, the detected strains were characterized by MST. Real-time PCR, targeting the IS1111 transposase, revealed the presence of Coxiella DNA in 15 cattle milk samples, in the brain, spleen, and lung of an aborted goat fetus, and in four goat milk samples. Real-time PCR, as already demonstrated for other important diseases (Lorusso et al. 2007, Polci et al. 2007, Lorusso et al. 2010, Pascucci et al. 2011), is a well-established, specific, and efficient molecular tool for direct diagnosis, thus circumventing the use of serological tests that often lack of sensitivity. Because the IS1111 transposase is a multicopy element, it represents a very sensitive target for a real-time PCR assay. Moreover, by means of quantitative real-time PCR, we defined the magnitude of shedding of C. burnetii in 19 milk samples. The combined results of this study confirm what has been previously discussed (Magnino et al. 2009); indeed, the bacterial excretion ranges from 102 to 105 bacteria/mL. The identification of heavy-shedder animals is crucial for the limitation of the infection to both animals and humans (Guatteo et al. 2007, Rodolakis et al. 2007). In this study, we identified 73,7% (14/19) of heavy-shedder cows (Guatteo et al. 2007) showing high to very high excretion of the pathogen through milk.
Typing all C. burnetii strains for epidemiological and management purposes is also critical. The MST method was selected in this study for Coxiella genotyping. The main benefit of MST is represented by the fact that the analysis of the spacer sequences permits us to understand the evolution of bacteria. Indeed, the spacers are more variable than multilocus sequence typing (MLST) targets housekeeping genes, because they are less susceptible to selection pressure and they are more stable than multilocus VNTR analysis (MLVA). Moreover, MST assay can be used “straight” on biological samples, thus avoiding isolation procedures that require biosafety level 3 (BSL3) facilities. Furthermore, the results can be easily compared to other global and localized studies that use MST as well (Glazunova et al. 2005, Hornsta et al. 2011, Pearson et al. 2014).
As already discussed (Tilburg et al. 2012), the amplification of COX56 and COX57 markers from biological samples presents some difficulties. For this reason a new forward primer for COX57 was designed, and the primer concentration combined with a different annealing temperature for COX56 and COX57 was adopted. These modifications generated the complete MST profile on milk and fetus samples.
The MST analysis showed the presence of a single MST profile in cattle (farm 1) and that the genotype circulating is different from all the sequence types present in the reference database but is related to isolates belonging to the ST20 that have been found in animal and human samples in the United Kingdom, France, Germany, Netherlands, Spain, Hungary, and United States. In particular, ST20 was associated most frequently with cattle and rarely with goats: A single goat sample was found in the Netherlands (Tilburg et al. 2012), in two milk samples in the United States (Pearson et al. 2014), and in a large goat herd in the United Kingdom in which it caused abortions (Reichel et al. 2012). However, in most cases, the genotype ST20 was associated with cattle and cow's milk (Glazunova et al. 2005, Astobiza et al. 2012, Pearson et al. 2014, Sulyok et al. 2014). Conversely, the sequence type detected in the aborted goat fetus (farm 3) was similar to ST8. This includes 35 strains (Hornsta et al. 2011), and it is one of the most represented genotypes in Europe. In fact, it has been found in France, Spain, and Portugal often associated with human cases with chronic clinical manifestations such as endocarditis (Glazunova et al. 2005, Astobiza et al. 2012, Santos et al. 2012). In the United States, this sequence type was reported recently in goat samples associated with human outbreaks (Kersh et al. 2013, Pearson et al. 2014).
ST8 may cause asymptomatic infections that remain undiagnosed (Hornsta et al. 2011), and consequently untreated, leading in some susceptible people to the development of endocarditis, vascular infections, or other less frequent symptoms of chronic Q fever. Monitoring farm animals and environment may represent a strategy to preserve the human population from exposure to strains of this MST group particularly prone to cause chronic disease.
Moreover, we make evident by the analysis of the goat milk sample collected from farm 2 a combination of alleles that generates a novel MST group. Remarkably, the profile differs significantly from all previously described genotypes including ST27, the closest sequence type. The results suggest the presence in the Abruzzo region of novel genotypes. None of them was related to ST33 involved in the Q fever outbreak in The Netherlands (Tilburg et al. 2012). The presence of different genotypes may be explained by the movements of cattle, sheep, and goats from other countries, where the information about the presence of Q fever is sporadic and studies on the genetic characterization of C. burnetii strains have never been attempted. A global network focused on the molecular epidemiology of C. burnetii is therefore warranted.
Conclusion
In conclusion, this study was designed to provide new insights into the molecular epidemiology of C. burnetii in Italy. Molecular characterization of the field strains was performed by MST, and three novel genotypes were identified. Ideally, the MST assay should be combined with MLVA, which, unlike MST, analyzes rapidly evolving markers. This combination of assays will help to further characterize strains belonging to the same MST group.
Footnotes
Acknowledgments
We deeply thank Dr. Alfreda Tonelli for English revision of the manuscript. Dr. Massimiliano Orsini and Dr. Iolanda Mangone are also thanked for the phylogenetic analysis. Funding was provided by the Italian Ministry of Health. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the Istituto Zooprofilattico Sperimentale dell'Abruzzo e del Molise.
Author Disclosure Statement
No competing financial interests exist.