
Abstract
Zika virus (ZIKV) is an emerging pathogen belonging to the Spondweni serocomplex within the genus Flavivirus. It has been isolated from several mosquito species. Two lineages of ZIKV have been defined by polyprotein homology. Using high-throughput sequencing, we obtained and characterized three complete genomes of ZIKV isolated between 1976 and 1980 in the Central African Republic. The three viruses were isolated from two species of mosquito, Aedes africanus and Ae. opok. Two sequences from Ae. africanus had 99.9% nucleotide sequence identity and 100% amino acid identity, whereas the complete genome obtained from Ae. opok had 98.3% nucleotide identity and 99.4% amino acid identity with the other two genomes. Phylogenetic analysis based on the amino acid sequence of the polyprotein showed that the three ZIKV strains clustered together but diverged from all other ZIKV strains. Our molecular data suggest that a different subtype of West African ZIKV strains circulated in Aedes species in Central Africa.
Introduction
Z
ZIKV was first isolated in April of 1947 from a febrile rhesus monkey placed in a cage close to the Zika Forest near Lake Victoria in Uganda. A few months later, at the same site, the same virus was isolated from an Aedes africanus mosquito (Dick et al. 1952). Subsequently, ZIKV has been isolated from other mosquito species, including Ae. apicoargenteus caught in the Zika Forest (McCrae and Kirya 1982), Ae. luteocephalus in Nigeria in 1969 (Fagbami 1979) and Ae. vitattus, Ae. furcifer, and Ae. aegypti in Côte d'Ivoire in 1999 (Akoua-Koffi et al. 2001). Like some other flaviviruses, such as those that cause yellow fever, dengue, and St. Louis, West Nile, and Japanese encephalitis, ZIKV is transmitted by mosquitoes. It is found in both Africa and Asia. Several seroprevalence studies have confirmed that nonhuman primates and other animals can be reservoir hosts of ZIKV (Darwish et al. 1983) and maintain the monkey–mosquito transmission cycle (Wolfe et al. 2001). In this sylvatic transmission cycle, humans are incidental hosts.
The first human case of ZIKV infection was described in 1964 in Uganda (Simpson 1964). In humans, ZIKV causes a febrile illness with symptoms characteristic of dengue fever, with a rash, fever, joint and muscle pain, headache, and periorbital pain (Simon et al. 2011). Serological studies also indicate widespread distribution of ZIKV in several Asian countries. The first major outbreak of human ZIKV infection was reported in 2007 on Yap and surrounding islands in the Federated States of Micronesia (Duffy et al. 2009). Recently, human cases were reported in travellers returning to the western hemisphere from Senegal (Foy et al. 2011), in 2010 in Cambodia (Heang et al. 2012), in 2013 and 2014 in French Polynesia (Musso et al. 2014), and in 2013 in New Caledonia (NATHNAC 2014).
On the basis of NS5 homology, three lineages of ZIKV can be discerned—an East African lineage, a West African lineage, and a more distantly related Yap lineage. Several full genome sequences of ZIKV are available in GenBank, such as the prototype sequence obtained from the strain isolated from the monkey in Zika Forest in 1947, a sequence corresponding to the Yap outbreak, and another to a human case in Nigeria in 1968. The aim of this study was to sequence and characterize the complete genomes of three strains of ZIKV isolated from two species of mosquito, Ae. africanus and Ae. opok, collected in 1976 (ARB 7701), 1979 (ARB 13565), and 1980 (ARB 15076) in the Central African Republic (CAR). Ae. africanus and Ae. opok are found mainly in areas of gallery forest and vegetation surrounded by banana plantations or raffia (Ngoagouni et al. 2012).
Materials and Methods
Mosquitoes were collected in July, 1976, November, 1979, and August, 1980, close to the city of Bozo. Those belonging to the same species were grouped, homogenized, filtered, and inoculated into the brains of newborn mice for amplification (three serial passages). A brain biopsy was performed on each dead mouse, and the sample was lyophilized and stored in closed glass vials at room temperature. Each strain was then inoculated onto Vero E6 cells (one passage), and serological characterization was performed on pellet cells. An immunofluorescence assay indicated the presence of ZIKV, which was confirmed by seroneutralization at the World Health Organization (WHO) collaborating reference center for research on arborviruses and hemorrhagic fever viruses at the Institut Pasteur de Dakar. For molecular characterization, RNA was extracted from the supernatant of infected VeroE6 cells or directly from brains of infected newborn mice with a QIAmp Viral RNA Mini Kit (Qiagen) according to the manufacturer's instruction. Extracted total RNA was treated with Turbo DNAse (Life Technologies) to remove all of the Mus musculus DNA genome and then retrotranscribed into cDNA with SuperScript III reverse transcriptase and random hexamer primers (Life Technologies).
The cDNA generated was amplified with Phi29 enzyme, as described previously (Berthet et al. 2008). Amplified DNA was quantified in the Quant-iT assay provided by Invitrogen, and a fixed amount of amplified DNA was sequenced with an Illumina® HiSeq 2000 Sequencer. A total of 5×106 reads with 100 bases were obtained for each sample. All reads were filtered according to quality, and those corresponding to the mouse genome sequence were removed with Bowtie 2.0 software with the M. musculus Mn10 sequence as reference. The viral reads corresponding to the ZIKV genome were selected by a similarity approach with BLASTN and BLASTX search tools and the three complete sequences of ZIKV available in GenBank. For each selected read, only the region that matched the viral genome was considered. All reads were initially assembled with ABYSS software with different values of k, and the contigs were then assembled into a “super assembly” with the CAP3 program to obtain the full-length viral genome.
The complete genomes obtained were aligned with the CLUSTALX algorithm in Geneious software v. 6.1.3 at the nucleic acid and amino acid levels, with manual editing to increase the quality of the alignment.
Results and Discussion
Totals of 36,286, 646,560, and 1,433,246 reads, representing 2.8%, 53.8%, and 62.5% of all reads, were obtained for the three strains ARB 7701, ARB 15076, and ARB 13565, respectively. The average coverage depth of each genome was 341× for ARB7701, 6060× for ARB15076, and 13,417× for ARB1356. One contig for each ZIKV strain was obtained after this double-assembly process, corresponding to the whole length of the viral genome. The three complete sequences of ZIKV were submitted to GenBank and registered under the accession numbers KF268948, KF268949, and KF268950.
Genomic analysis of our ZIKV variants showed the classical organization of flaviviruses, with a polyprotein encoding three structural genes and seven nonstructural genes and two noncoding regions located at the 5′ and 3′ends of the viral genome. The organization of the open reading frame of all strains was conserved in comparison with other ZIKV genomes. In two genomes (ARB 7701 and ARB 13565), an addition of six amino acids was observed in the glycoprotein genes, starting at position 445 based on the polyprotein sequence.
The complete sequences of the two strains of ZIKV isolated from Ae. africanus (ARB 7701 and ARB 13565) had 99.9% sequence identity and 100% amino acid identity, whereas the third sequence, isolated from Ae. opok (ARB 15076), had 98.3% identity and 99.4% amino acid identity with the other two sequences. All of the sequences have an amino acid identity of 96.8–98.3% with sequences of ZIKV isolated in Micronesia (EC Yap), Nigeria (IbH 30656), and Uganda (MR 766) (Table 1). A high level of identity was observed in this study, although this was not shown yet for ZIKV. In Holmes' study, it has been shown that the arboviruses (alphaviruses and flaviviruses) have strong evolutionary constraint on their genomes and therefore evolve slowly (Holmes 2003). Nevertheless, in the two serotypes Spondweni and Zika, there was a significant diversity between the ZIKV species, with up to 25.4% divergence at the amino acid level along the polyprotein.
Normal type, percentage nucleotide identity; boldface type, percentage amino acid identity.
CAR, Central African Republic.
The phylogenetic analysis based on the Bayesian method with the nucleotide sequence of the complete genome available from GenBank showed that the four ZIKV strains isolated in CAR from Ae. africanus and Ae. opok clustered together but diverged from all other strains (Fig. 1). The three strains of ZIKV (ARB 7701, ARB 13565, and ARB 15076) isolated in the CAR came from the same region, close to Bozo, in tropical forest in the southwest but were sampled from mosquitoes at different times, the first being obtained in July, 1976 (ARB 7701) and the others in November, 1979 (ARB 13565) and August, 1980 (ARB 15076). Our data indicate that the two strains isolated from Ae. africanus evolved minimally during the 3 years between capture of mosquitoes. The difference in clustering between these ZIKV strains was explained by the presence of an additional sequence of six amino acids in the E glycoprotein of the strains isolated from Ae. africanus (at position 445–450 of the polyprotein). This deletion at a potential glycosylation site has already been observed in several other strains (Haddow et al. 2012), especially in the E protein of MR766 strain (acc. no. DQ859059). Studies on this glycosylation motif (N-Y-T/S) at residues 154–156 associated glycosylation with the ability of the West Nile virus strain to cause significant human outbreaks (Shirato et al. 2004). The strain isolated during an outbreak in Micronesia contains the complete site of N-glycosylation with the motif N-D-T; the majority of the other strains harbor this motif, due to either complete deletion of several amino acids or mutations.
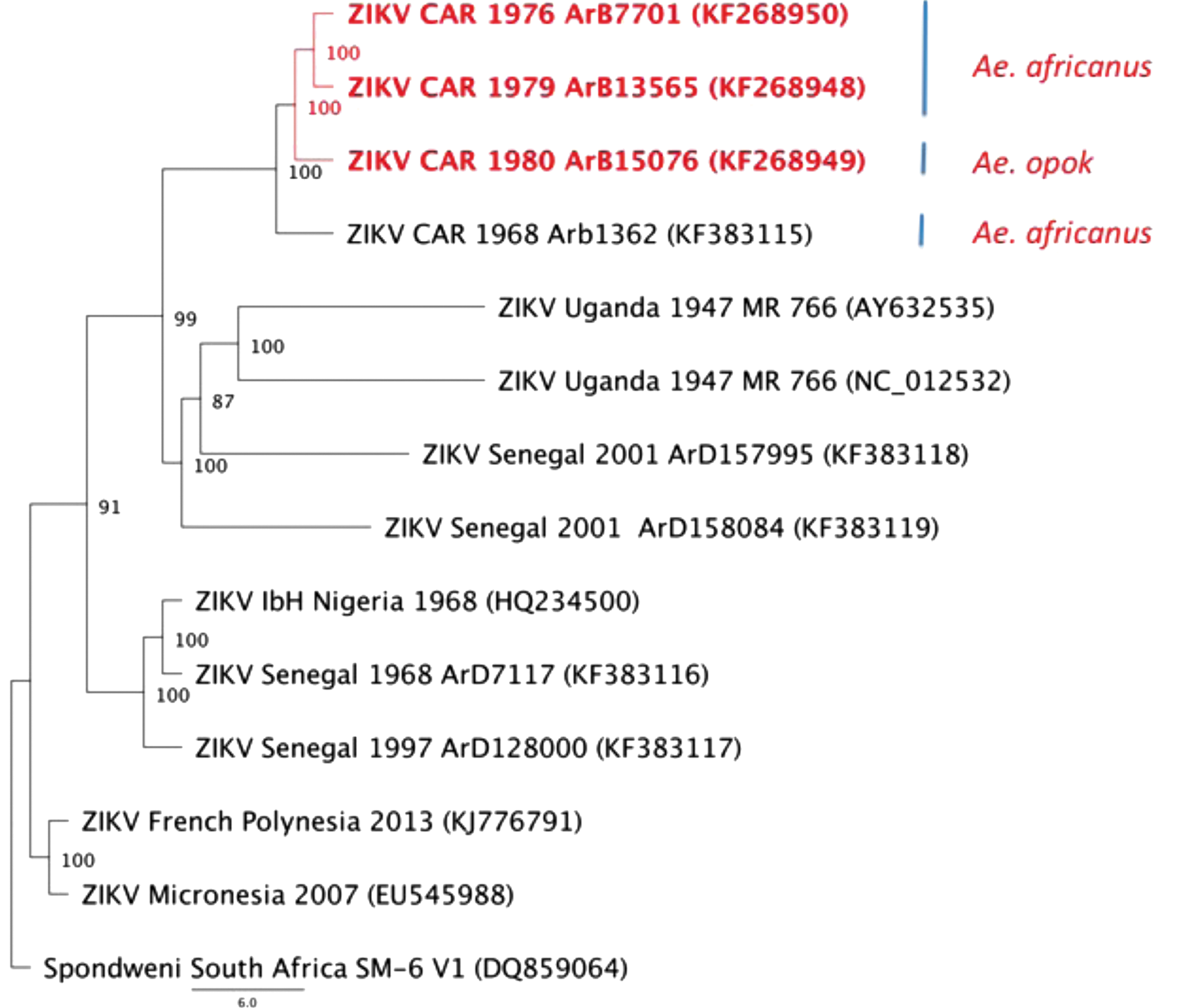
Phylogenetic tree of flavivirus belonging to the Spondweni group. Phylogenetic analysis of the complete genome was analyzed with sequences available in GenBank and were performed with MrBayes V.3.2 software, using the default chain for two million generations with the GTR+G+I nucleotide substitution model. Scale bar indicates nucleotide substitutions per site (Drummond et al. 2007).
Serological evidence has confirmed ZIKV circulation in humans in several countries of Central Africa (CAR, Egypt, Gabon, Sierra Leone, Uganda, and the United Republic of Tanzania), although only one strain has been isolated from a human case in CAR (Robin and Mouchet 1975, Jan et al. 1978, Saluzzo et al. 1981, Adam et al. 2005). These data suggest potential circulation of this virus through a sylvan natural cycle in the Congo basin forest. Our molecular data show that ZIKV strains isolated in CAR evolved from an ancestral strain belonging to the African lineage; however, these strains seem to differ from other groups, particularly from the strain recently isolated in Gabon (Grard et al. 2014). Detection of ZIKV in Ae. albopictus in Gabon demonstrates the ability of this virus to adapt to a novel vector.
In conclusion, the recent introduction of Ae. albopictus in CAR suggests potential emergence of ZIKV in different competent mosquitoes (Kamgang et al. 2013). Viral surveillance should be set up in Central Africa to ensure early detection of the spread of ZIKV in the human population.
Footnotes
Acknowledgments
This study was supported by the Programme Transversal de Recherche CEVACAR No. 385 financed by the Institut Pasteur (Paris, France). The funder had no role in study design, data analysis, or preparation of the manuscript.
Author Disclosure Statement
No conflicting financial interests exist.
N.B., E.N., A.G., J.C.M., and M.K. conceived and designed the experiments. N.B. and B.S. performed the experiments. N.B. and S.D.D. performed the bioinformatics analysis. N.B., A.G., J.C.M., and M.K. analyzed the data. N.B., B.K., J.C.M., and M.K. wrote the manuscript.