
Abstract
Mosquito-borne alphaviruses have the potential to cause large outbreaks throughout the world. Here we investigated the causative agent of an unexpected Sindbis virus (SINV) outbreak during August–September, 2013, in a previously nonendemic region of Sweden. Mosquitoes were collected using carbon dioxide–baited CDC traps at locations close to human cases. The mosquitoes were initially screened as large pools by SINV-specific quantitative RT-PCR, and the SINV-positive mosquitoes were species determined by single-nucleotide polymorphism (SNP) analysis, followed by sequencing the barcoding region of the cytochrome oxidase I gene. The proportion of the collected mosquitoes was determined by a metabarcoding strategy. By using novel strategies for PCR screening and genetic typing, a new SINV strain, Lövånger, was isolated from a pool of 1600 mosquitoes composed of Culex, Culiseta, and Aedes mosquitoes as determined by metabarcoding. The SINV-positive mosquito Culiseta morsitans was identified by SNP analysis and sequencing. After whole-genome sequencing and phylogenetic analysis, the SINV Lövånger isolate was shown to be most closely similar to recent Finnish SINV isolates. In conclusion, within a few weeks, we were able to detect and isolate a novel SINV strain and identify the mosquito vector during a sudden SINV outbreak.
Introduction
O
SINV is an enveloped, positive-sense, single-stranded (ss) RNA virus, divided into five genotypes on the basis of genome sequences. Interestingly, strains from northern Europe and South Africa have been found to be related and grouped together in a genetic subcluster—the African–European genotype (genotype 1) (Strauss et al. 1984, Lundström and Pfeffer 2010).
In Sweden, SINV causes a disease known as Ockelbo disease, Pogosta disease in Finland, and Karelian fever in Russia, all synonyms for the same disease (Lvov et al. 1988). The major symptoms are joint inflammation, rash, fever, fatigue, and headache (Kurkela et al. 2005, Sane et al. 2012a). The joint symptoms may persist for months or even years (Skogh and Espmark 1982, Niklasson and Espmark 1986, Niklasson et al. 1988, Kurkela et al. 2008). The first cases of Ockelbo disease in Sweden were reported in the 1960s, and later this disease was shown to be associated with a strain of SINV isolated from mosquitoes in Edsbyn, Sweden (Skogh and Espmark 1982, Niklasson et al. 1984). Cases of Ockelbo disease are frequently diagnosed in central Sweden (Espmark and Niklasson 1984, Lundstrom et al. 1991), but recently also in northern Sweden (Ahlm et al. 2014). In Sweden, SINV has been isolated from ornithophilic mosquitoes Culex (Cx.) pipiens and/or Cx. torrentium and Culiseta morsitans, as well as from the bridge vector Aedes (Ae.) cinereus (Francy et al. 1989). Moreover, Cx. torrentium has also been shown to transmit the virus to susceptible hosts (Lundstrom et al. 1990, Lundstrom 1994). The circulation of these and other possible mosquito vectors has recently been determined in Sweden (Lundström et al. 2013), and their distribution may have widened due to global warming (Gould and Higgs 2009, Lindgren et al. 2012). It is still not known whether SINV circulates constantly in endemic areas or whether outbreaks are caused by reintroduction of new viral strains by, for example, passerine birds.
In August and September of 2013, an outbreak of Ockelbo disease occurred in northern Sweden, along the coastline of the Gulf of Bothnia (Fig. 1), north of the endemic region where SINV is usually found. By using novel techniques (Engdahl et al. 2014) and a high-throughput screening assay for virus detection, we were able to isolate and characterize the disease-causing virus within a short timeframe from an area where numerous human cases had occurred. After complete genome sequencing of this novel northern Swedish isolate, its evolutionary relationship to other SINV strains was investigated.
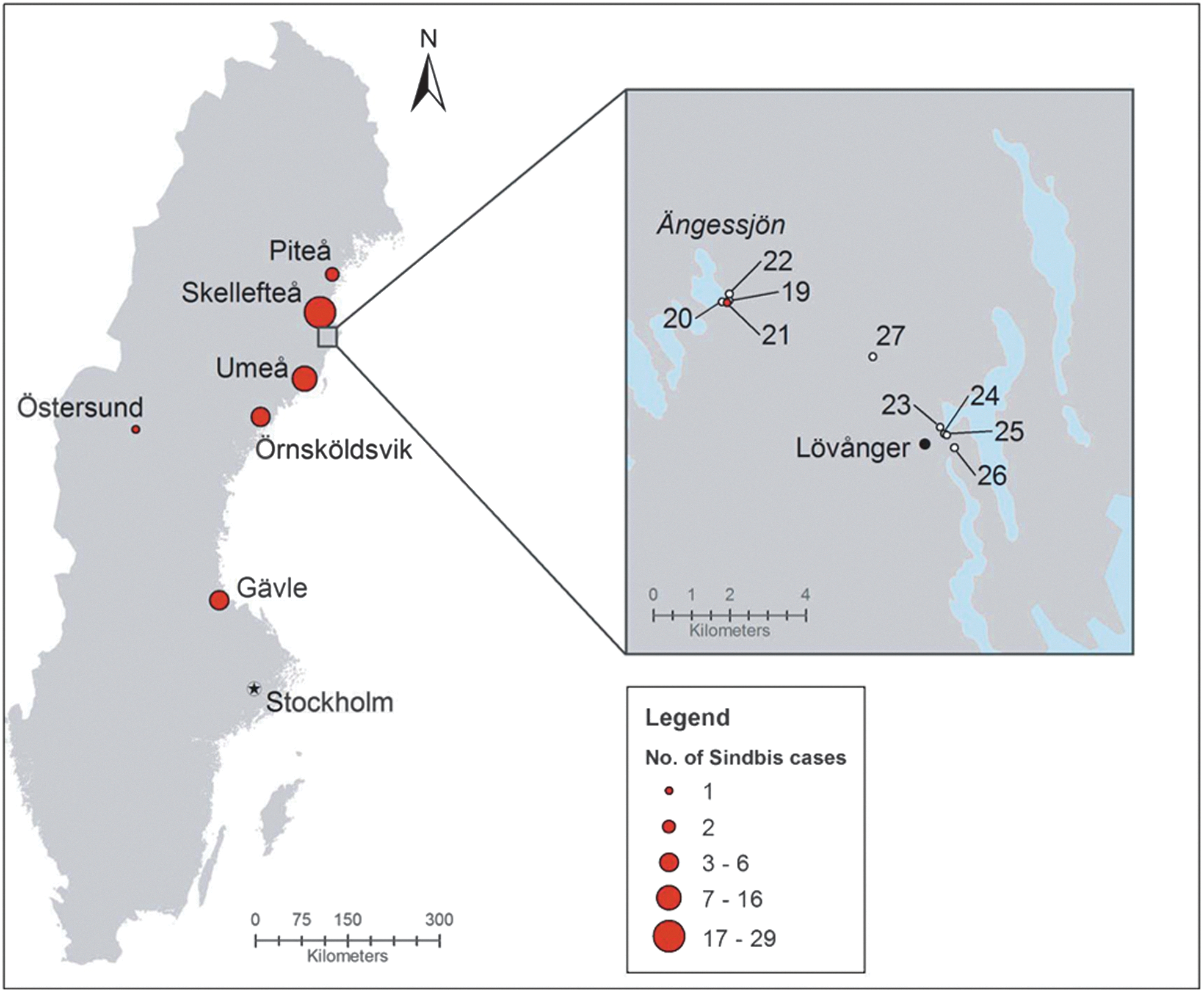
Mosquito collection sites. This map shows the location and approximate numbers of individuals who tested positive for Sindbis virus (SINV) antigens during August–September, 2013. The mosquito capture sites are indicated in a magnified map at the upper right side of the figure. Four CDC traps (points 19–22) were placed near the lake “Ängessjön” outside the village of Lövånger between September 6 and September 13. This site is a wetland and bird-nesting area at the geographic position N64.40 E21.20. Four other traps (points 23–26) were placed near the village. This site is a mixed forest area at location N64.37 E21.31. Trap point 27 was at slightly higher altitude, along a creek in a mixed forest area at position N64.39 E21.28. Trap point 21 was the position where the SINV-positive mosquitoes were collected. Color images available online at
Materials and Methods
Mosquito collection
Mosquitoes were collected at locations where Ockelbo disease patients might have been infected. Nine carbon dioxide–baited CDC traps were placed in the vicinity of the village Lövånger, in Västerbotten County, Sweden (Fig. 1), and 1600 mosquitoes were captured between September 6 and September 13. The traps were placed 1.5–2 meters above ground in the late afternoon, and the mosquitoes were collected the following morning.
Sample preparation (homogenization, incubation, RNA extraction, and pooling)
RNA yields from captured mosquitoes were evaluated using different homogenization procedures, bead materials, buffers, and incubation times before RNA isolation. Briefly, individual mosquitoes were placed in 2-mL microcentrifuge tubes with five 2-mm diameter stainless steel beads (Retsch GmbH, Germany) and 350 μL of cell culture medium (Glasgow MEM; GIBCO, Invitrogen, Carlsbad, CA). The homogenization was performed at 4°C with a Mixer Mill 400 (Retsch GmbH, Germany) for 20 s at a relative frequency of 30 cycles per second.
To evaluate and optimize sample preparation procedures, in vitro–transcribed RNA was spiked with uninfected Ae. aegypti mosquitoes and quantified by PCR. The in vitro–transcribed RNA was produced and analyzed as described previously (Naslund et al. 2008, Mohamed et al. 2013). From individual mosquito homogenates, 25-μL aliquots from 10 mosquitoes were pooled to 10× pools. Furthermore, from ten 10× pools, 25-μL aliquots were taken to make 100× pools and subsequently 1000× pools (Fig. 2). Samples containing 100 μL of homogenate were prepared for RNA extraction by incubation for 30 min at 37°C in 0.2% (wt/vol) sodium dodecyl sulfate (SDS) and 50 μg/mL proteinase K before RNA isolation with QIAmp Viral RNA (Qiagen, Hilden, Germany). Screening for viral RNA was performed as indicated below, and samples that were PCR-positive for SINV were traced back by sequential screening of 1000× and 100× pools to 10× pools to individual samples.
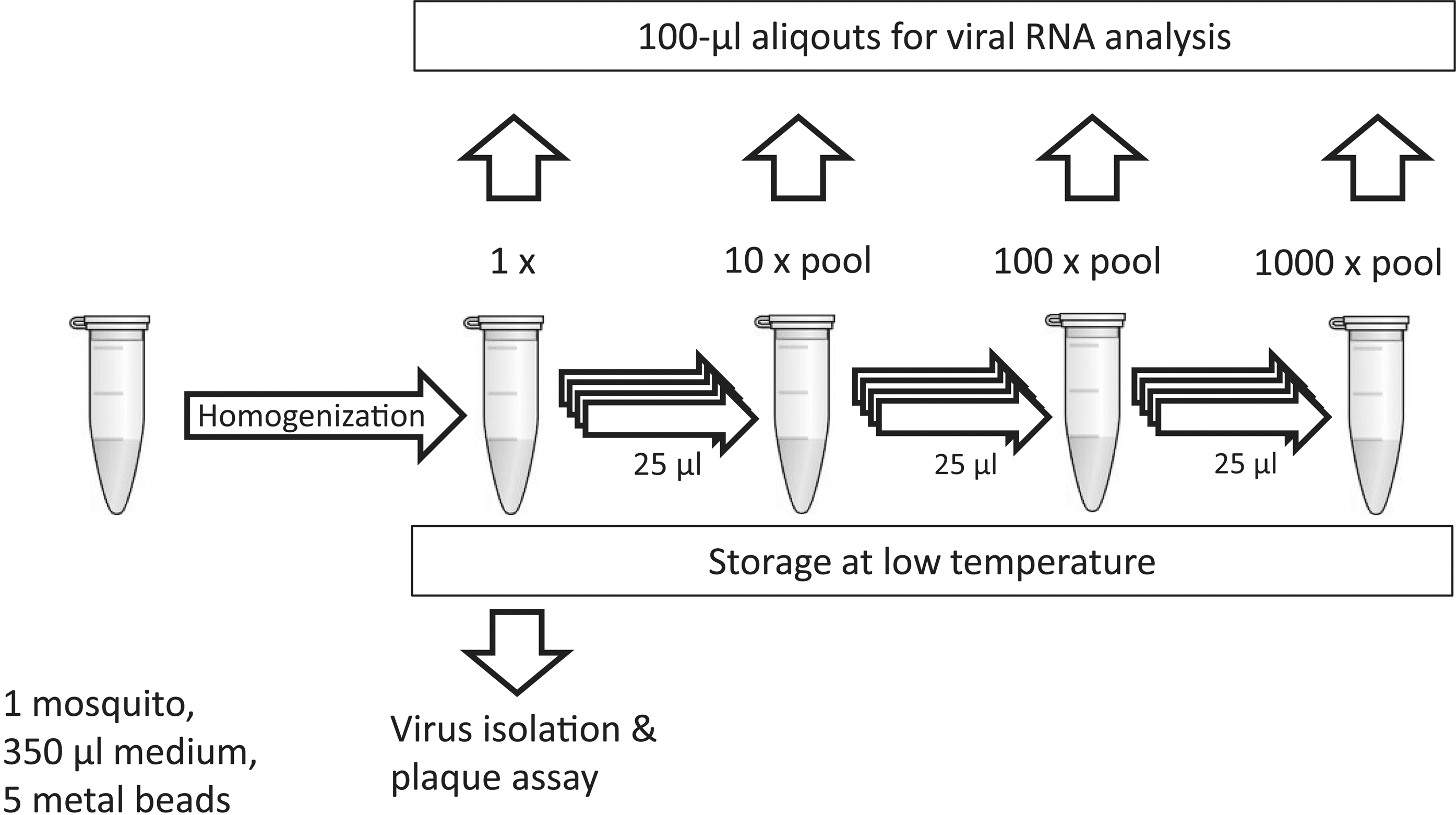
Sequential pooling and screening procedure. This illustrates how the captured mosquitoes were homogenized and how samples were prepared and analyzed individually and as pools containing 10, 100, and 1,000 mosquitoes. Aliquots of these pools were then used for RNA isolation and stored at −80°C until to await virus isolation and plaque assay of PCR-positive samples.
Screening (PCR cycling, primers)
SINV RNA screening was performed by qRT-PCR amplification using the CFX96TM Real-time Detection System (Bio-Rad Laboratories, Hercules, CA). Reverse transcription prior to SYBR Green– or probe-based qRT-PCR amplification was carried out with 0.4 μL of Superscript III (Invitrogen, Carlsbad, CA) and 10 μL of 2× master mix for 5 min at 42°C, or 10 min at 50°C, respectively.
For SYBR Green–based analysis, KAPA SYBR FAST One-Step qPCR Kits were used (KAPA Biosystems, Woburn, MA). The amplification was carried out in 20-μL reaction mixtures containing 7.6 μL of template and SinF (5′-GTGAGCACCCCGCACAAAAATGAC-3′) and SinR (5′-GCCAAAAAGGGCAAACAGCCAACTC-3′) primers (200 nM each).
For probe-based detection, the qSCRIPT XLT One-Step RT qPCR ToughMix (Quanta Biosciences, Gaithersburg, MD) was used as above, but with 5.5-μL template, SinFP/SinRP primers (5′-GGTTCCTACCACAGCGACGAT-3′ and 5′-TGATACTGGTGCTCGGAAAACA-3′) at 400 nM each, and SinP probe (5′-FAM-TTGGACATAGGCAGCGCA-BHQ1-3′) at 125 nM (Sane et al. 2012b).
The cycling conditions for SYBR Green– and probe-based amplification were as follows: Denaturation at 95°C for 3 min followed by 40 cycles involving 3 s of denaturation at 95°C and 30 s of annealing/elongation at 60°C. Positive and negative controls were included in each test.
Identification of mosquito species using SNP analysis
SNP analysis was carried out essentially as described before (Engdahl et al. 2014). The species identity of the SINV-positive mosquito, as suggested by SNP analysis (Table S1; Supplementary Data are available at
Virus isolation
Aliquots of the homogenate from the SINV PCR-positive mosquito were diluted in Glasgow MEM and added to monolayers of BHK-21 cells (American Type Culture Collection [ATCC] number CCL-10). After allowing 90 min for virus attachment, the cells were washed and incubated with Glasgow MEM supplemented with 5% fetal calf serum (FCS), 1.3 grams/L tryptose (Difco™; Becton, Dickinson and Company, Sparks Glencoe, MD), 10 mM HEPES, 1 mM sodium pyruvate, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in 5% CO2. After 24–36 h, viral stocks were harvested by repeated freeze–thawing before being stored at −80°C.
Plaque assay
Monolayers of BHK-21 cells grown in six-well plates were infected, as above, with serially diluted mosquito homogenates or previously prepared viral stocks. For plaque assay, the wells were washed twice with cell culture medium, covered with 3 mL of 1.2% carboxymethyl cellulose (CMC) (Aquacide II; Calbiochem®, CA) in Dulbecco's modified Eagle medium (DMEM) (GIBCO) with 2.5% FCS, and incubated for 5 days at 37°C in 5% CO2. The cells were fixed for 10 min by addition of 5 mL of 10% formaldehyde in PBS and then washed with water before counterstaining with 1% Crystal Violet in 20% ethanol and 0.7% NaCl (Naslund et al. 2009). The wells were finally washed with tap water to visualize the plaques.
Sequencing and phylogenetic analysis
Available SINV sequences were downloaded from GenBank and primers were designed with the aim of amplifying and sequencing the complete viral genome (see Table S2). PCR amplification was performed as described above in standard 96-well plates using the KAPA SYBR® FAST qPCR Kit. The PCR products were purified on Illustra MicroSpin S-400 HR Columns (GE Healthcare, Sweden) and sequenced (Eurofins MWG Operon, Ebersberg, Germany).
Phylogenetic placement of the SINV Lövånger strain was performed using the maximum likelihood method and the TIM2-I-G model based on partial polyprotein gene sequences for SINV strains of different genotypes. The phylogenetic model was selected based on the Bayesian information criteria (BIC) using Jmodeltest v.2.1.5 (Darriba 2012), and the phylogenetic tree was inferred using phyml v. 3.1. The reliability of the tree was tested using nonparametric bootstrapping based on 500 pseudoreplicates.
The NeighborNet method was used for local phylogenetic positioning of the SINV Lövånger strain within the SINV genotype I group using two datasets. The first dataset contained 12 whole-genome sequences and the second 13 partial polyprotein gene sequences. Phylogenetic networks were inferred using Splitstree v. 4.12.8 (Huson and Bryant 2006) and the NeighborNet method (Bryant and Moulton 2004) with an uncorrected-p distance model. Sites containing gaps were excluded. Weights were used in the EqualAngle transformation. All phylogenetic analyses were based on sequence data that were aligned using Muscle v.3.8.31 (Edgar 2004). Strain designations and accession numbers are provided in the figures.
Metabarcoding
A reference database for Swedish mosquitoes was created by Sanger sequencing of COI gene fragments of morphologically identified mosquitoes along with COI gene sequences from GenBank. To identify the frequency of mosquito species in the 1600 mosquitoes collected, two pooled samples were prepared representing the two capture periods, and DNA was amplified using four primer combinations; LCO1490+HCO2198; GB1310_29F+GB_1960_1936R; GB_1358_83F+TL2-N-3014R; and GB1310_29F+TL2-N-3014R (Folmer et al. 1994, Engdahl et al. 2014). The resulting fragments were then processed for Illumina sequencing with the Nextera XT Kit (Illumina Inc.) and a bioanalyzer, indicating fragment lengths of 500–700 bp, and subsequently sequenced with an Illumina Miseq using the 300+300 bp V3 kit (Illumina Inc.).
The sequence reads produced were compared to the mosquito database with BLASTn. Top hits longer than 300 bp, with more than 98% identity to the reference, were counted, and the proportion of reads matching each species were taken as a measure of relative abundance of each species in the mosquito pool.
The sequence reads from each primer fragment were also combined and used for de novo assembly of contigs by using mira4 (Chevreux 1999, Chevreux 2004). This assembly also takes the paired reads into account and provides longer sequences for comparison than single reads matched against the database. The contigs obtained were finally compared to the reference database with BLASTn. Finally, contigs matching specific species were counted and proportions of reads matching each species were taken as a measure of relative abundance of each species in the pool.
Results
SINV-positive mosquitoes were detected by high-throughput RT-PCR screening
Mosquitoes were captured in the vicinity of Lövånger over two consecutive sampling rounds and resulted in a total collection of 1600 mosquitoes. After sample preparation followed by screening using SINV-specific RT-PCR, one pool of 1000 and one pool of 100 showed strong PCR signals, by both SYBR Green– and probe- based amplification and detection. After successively tracing back to individual mosquito samples, one highly positive mosquito that was collected on September 8, 2013, was identified.
To estimate the sensitivity of the assay further, the positive 1000× pool was diluted 10 times further with a sample prepared from SINV-negative Ae. aegypti mosquitoes, resulting in a pool of 10,000 mosquitoes. Even this highly diluted pool showed a clear amplification signal by both qRT-PCR methods.
Type-specific identification of the mosquito collection and the SINV-positive mosquito
By using a previously published DNA barcoding typing method (Engdahl et al. 2014), we were able to identify the species of the SINV PCR-positive mosquito by mismatch amplification mutation assay (Melt-MAMA analysis) (Birdsell et al. 2012, Engdahl et al. 2014). The mosquito species was determined through a hierarchical combination of PCR reactions (Fig. 3). After sequencing followed by comparisons to COI gene sequences in GenBank, the SINV-positive mosquito species was determined to be C. morsitans.

Identification of mosquito species. This figure shows the algorithm for species identification of the Sindbis virus (SINV)-positive mosquito through sequential examination by single-nucleotide polymorphism (SNP) analysis and melting point analysis of the resulting PCR products. P1, P7, P8, and P9 were the preferred primer pairs (see Table S1). From this algorithm, shown by arrows, the species of the SINV-positive mosquito was determined.
To estimate the proportion of different mosquito species in the sampled area, a metabarcoding approach was used where DNA extracted from pools of collected mosquitoes was amplified and sequenced and the proportion of different species in the sample was estimated. Using mosquito pools of known proportions the GB1310_29F+GB_1960_1936R primer combination underestimated the proportion of C. morsitans, whereas the GB_1358_83F+TL2-N-3014R primer pair underestimated the proportion of Ae. hexodontus, Ae. punctor, and Ae. punctodes (data not shown). To circumvent this bias, all sequence reads from the different amplification products were pooled and assembled de novo into contigs producing longer sequences that could be matched to the database. These results showed that Cx. pipiens and C. morsitans together with mosquitoes from the punctor group (Ae. hexodontus/Ae. punctor/Ae. punctodes), the annulipes group (Ae. cantans/Ae. annulipes/Ae. excrucians) and the subgenus Aedes (Ae. cinereus/Ae. rossicus) were the major species among the 1600 mosquitoes collected.
Isolation of the SINV Lövånger strain of SINV
When the mosquito homogenate from the SINV-positive mosquito was added to BHK-21 cells, a strong cytopathogenic effect was observed. The titer of the virus stock obtained was estimated by plaque assay to be 1.5× 108 pfu/mL. The mosquito homogenate was analyzed at the same time and the complete sample was estimated to contain 3.5× 104 pfu (data not shown), which corresponded to a freshly prepared sample with 1× 106 pfu (data not shown).
Phylogenetic analysis of the SINV Lövånger strain of SINV
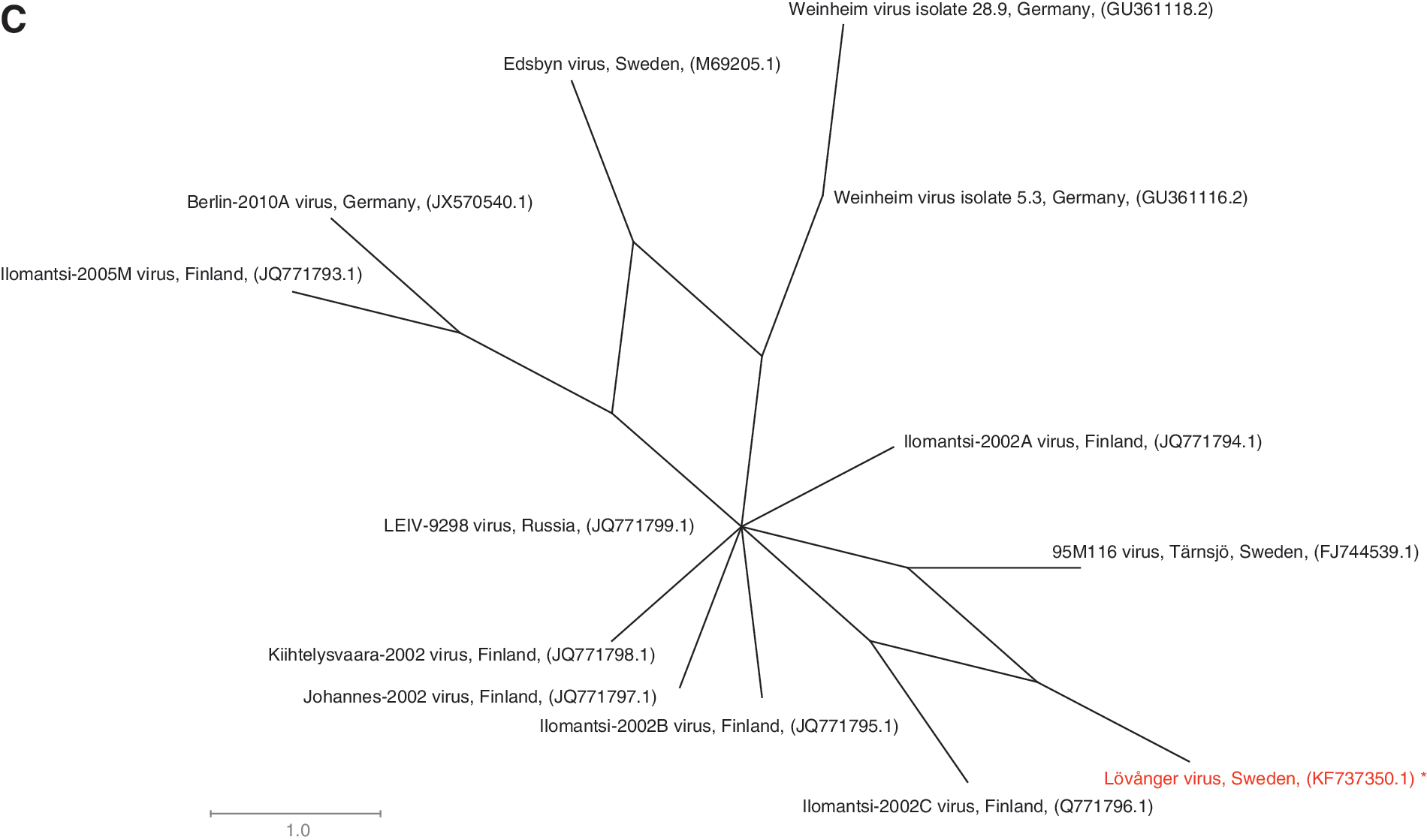
By maximum likelihood phylogeny based on partial polyprotein sequences of SINV strains of known genotypes (2110 shared nucleotide positions), the SINV Lövånger strain clusters with strains of SINV genotype I (Fig. 4A). Due to poor reliability of the phylogeny for strains of genotype I, as indicated by low bootstrap values, the phylogenetic position of the Lövånger strain within the SINV genotype I group was investigated further by the NeighborNet method. This method highlights homoplasy in the data by inference of a network structure. When we analyzed whole-genome sequences for closely related SINV genotype I strains (11,518 shared nucleotide positions), the SINV Lövånger strain was found to most closely resemble the SINV Ilomantsi-2002C strain, which was isolated in Finland (Fig. 4B), with no phylogenetic conflicts. When the same dataset was expanded with the SINV 95M116 strain from Tärnsjö, Sweden, for which only 2188 nucleotide positions of the polyprotein gene are available, homoplastic signals became evident for the SINV strains Ilomantsi-2002C, Lövånger, and 95M116 (Fig. 4C). The true branching order for these Swedish and Finnish strains therefore remains unclear.

Phylogeny.
Discussion
We have described the isolation and characterization of a disease-causing virus during an unexpected and large outbreak of Ockelbo disease in a previously nonendemic area of northern Sweden. Human disease has mainly been reported from northern Europe and South Africa, but intriguingly, human SINV cases are not reported in Central Europe despite the fact that SINV can be detected in birds and mosquitoes (Jost et al. 2010, Jost et al. 2011, Eiden et al. 2014). The reason for this is unknown, but increased awareness and surveillance for SINV infection would be of importance.
After screening mosquitoes for SINV RNA, we isolated SINV from the ornithophilic mosquito C. morsitans, a known SINV vector (Francy et al. 1989). Furthermore, we applied a recently developed technique for genetic typing of the SINV-positive mosquito by SNP analyses of the COI gene (Engdahl et al. 2014).
A new SINV strain (Lövånger) was isolated, and the full genome was sequenced. As expected, this strain was found to cluster with strains of SINV genotype I in a maximum likelihood tree. To date, isolates belonging to SINV genotype I mainly originate from South Africa, Sweden, Finland, and Germany (Lundström and Pfeffer 2010, Storm et al. 2013). When the entire SINV genome was sequenced, the Lövånger strain clustered with the Finnish strain Ilomantsi-2002C. By inclusion of the partial polyprotein gene of the Swedish strain 95M116, the SINV Lövånger strain was found to contain homoplastic SNPs linking it both to the Finnish Ilomantsi-2002C strain and the Swedish 95M116 strain, but without the possibility of resolving their phylogenetic relationships. Incompatible nucleotide sites were also detected among other strains (e.g., Edsbyn, Ilomantsi-2005M, and Weinheim virus isolate 5.3), indicating that genetic reversals or convergent mutations are not infrequent in SINV. Introduction of new SINV strains by migratory birds could also be a possibility (Lundström and Pfeffer 2010). Although the levels of homoplasy detected in the data provide insight into the evolution of SINV, this finding also underscores the importance of full-length genomic sequences in investigation of genetic relationships among isolates.
By use of next-generation sequencing, an estimation of proportions of species in a pool of collected organisms can be made, because the number of reads has been shown to be proportional to the biomass of species present (Zhou et al. 2013). We used this technique to determine the species proportions of mosquitoes. The resulting proportions from morphologically identified mosquitoes from the same area indicated that the mosquito fauna was different from samples taken earlier (Engdahl et al. 2014). Interestingly, the proportion of Cx. pipiens and C. morsitans was unexpectedly high in our mosquito samples, compared to a previous collection of mosquitoes in this region (Engdahl et al. 2014), whereas Ae. communis was less frequent and the number of Ae. cinereus was similar in previous collections. Although we did not detect the known competent vector Cx. torrentium (Lundstrom et al. 1990), both C. morsitans and Cx. pipiens are known to be ornithophilic and could potentially transmit SINV in the enzootic cycle between mosquitoes and birds (Francy et al. 1989). It is possible that the virus was spread among birds by these ornitophilic mosquitoes and then passed on to humans, e.g., by Ae. cinereus. An interesting finding that needs to be studied further is if changes in prevalence of mosquito species, such as Culiseta morsitans and Cx. pipiens, could drive the transmission of SINV in local bird populations, eventually leading to an outbreak among humans.
Conclusions
In conclusion, during a recent outbreak of Ockelbo disease along the coast of northern Sweden, we isolated SINV from a mosquito collected at locations where high numbers of human cases occurred. Mosquitoes were screened for SINV using a high-throughput method developed by us, and we succeeded in isolating SINV from the ornithophilic mosquito C. morsitans. A new SINV strain (Lövånger) was isolated, and the full genome was sequenced. The strain was found to contain homoplastic SNPs that linked it to SINV strains from Ilomantsi, Finland, and Tärnsjö, Sweden. To improve our understanding of SINV epidemiology and factors triggering outbreaks, further studies of the molecular epidemiology are warranted. Such studies should include sampling of birds and mosquitoes during outbreaks, and even during interepidemic periods, and possibly also sampling of recognized human cases.
Footnotes
Acknowledgments
We thank Gustaf Granlund for suggesting the capture spots. We are grateful to Samuel Öhman for bioinformatics and to Per Wikström for graphics. The study was supported by the County Council of Västerbotten. The complete genome sequence of the Sindbis virus strain Lövånger is stored in GenBank, accession number KF737350.
Author Disclosure Statement
No competing financial interests exist.
Author contributions: J.B. prepared RNA samples, did PCR analysis, prepared samples for sequencing, and contributed to the writing. O.F. placed the traps and collected the mosquitoes. P.L. did the bioinformatics and contributed to writing. J.N. participated in the experimental design, preparation of samples, and the virus-titer determinations. T.L. performed next-generation sequencing, metabarcoding, and data analysis. C.E. contributed to the experimental design. A.L. contributed to the discussion of the results. Å.G. contributed with epidemiological surveillance and analysis of the geographic distribution of the epidemic. C.A. and M.E. organized the trapping and contributed to writing and proofreading of the manuscript. G.B. coordinated and participated in the experimental design, performed virus isolation, and drafted the manuscript. All the authors read and approved the final manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.