
Abstract
Candidatus Rickettsia asemboensis was identified molecularly in fleas collected in 2009 from Asembo, Kenya. Multilocus sequence typing using the 17-kD antigen gene, rrs, gltA, ompA, ompB, and sca4 demonstrated that Candidatus R. asemboensis is closely related to Rickettsia felis but distinct enough to be considered for separate species classification. Following this molecular characterization of Candidatus R. asemboensis, the in vitro cultivation of this bacterium was then performed. We used Ctenocephalides canis and Ctenocephalides felis fleas removed from dogs in Kenya to initiate the in vitro isolation of Candidatus R. asemboensis. Successful cultures were obtained using Drosophila melanogaster S2 and Aedes albopictus C6/36 cell lines. Cytological staining and quantitative real-time PCR (qPCR) assays were used to visualize/confirm the culture of the bacteria in both cell lines. Sequencing of fragments of the 17-kD antigen gene, gltA, and ompB genes confirmed the identity of our Candidatus R. asemboensis isolates. To date, we have passaged Candidatus R. asemboensis 12 times through S2 and C6/36 cells, and active and frozen cultures are currently being maintained. This is the first time that a R. felis–like organism has been grown and maintained in culture and is therefore the first time that one of them, Candidatus R. asemboensis, has been characterized beyond molecular typing.
Introduction
C
For full biological characterization of Candidatus R. asemboensis, isolation of the bacteria from fleas, and growth in vitro is necessary. In past studies, we have successfully used insect cells to isolate/propagate different rickettsial species, i.e., Candidatus R. andeanae (Luce‐Fedrow et al. 2012), R. felis (Luce-Fedrow et al. 2014), and R. africae (Mr. Teik-chye Chan, NMRC, pers. commun.). To isolate Candidatus R. asemboensis, we used C. canis and C. felis fleas collected from dogs in the Asembo area of Siaya County in western Kenya. The fleas were processed and used to infect the Aedes albopictus C6/36 cell line and the Drosophila melanogaster S2 cell line. We monitored and confirmed the identity of the rickettsiae growing in the cell lines by: (1) A species-specific qPCR assay for Candidatus R. asemboensis (Jiang et al. 2013), (2) Acridine Orange staining, (3) transmission electron microscopy, and (4) DNA sequencing. Our Candidatus R. asemboensis–infected cell lines have been maintained since July, 2013, and have been passed 12 times through June 2014.
Materials and Methods
Maintenance of cell lines
The Drosophila S2 cell line (Drosophila Genomics Resource Center; Bloomington, IN) and the A. albopictus C6/36 cell line (gift of Dan Ewing, Naval Medical Research Center, Silver Spring, MD) were cultivated at 25°C in Schneider's Drosophila medium (Gibco/Invitrogen, Carlsbad, CA) supplemented with 5% or 10% fetal bovine serum (FBS; heat-inactivated) (Atlanta Biologicals, Lawrenceville, GA). Antibiotics were not used for cell culture.
Flea collection and processing
This study was conducted in Asembo area of Siaya County in western Kenya. This is within the area covered by the ongoing Kenya Medical Research Institute/Centers for Disease Control and Prevention (KEMRI/CDC) population-based infectious disease surveillance (PBIDS), which has been in operation since early 2006. Study compounds were randomly selected from livestock-owning compounds (LOCs) from which the novel Rickettsia spp. Candidatus R. asemboensis, had been previously detected in cat and dog fleas (Jiang et al. 2013). Informed consent was sought from the household head to allow us to inspect the dogs, and the cats (when present) for the presence of fleas. Fleas were collected from dogs and cats and immediately transferred to a single 50-mL Falcon tube (VWR, Radnor, PA) per host. Every effort was made to ensure that the fleas remained alive until the time they were delivered to the field lab. The GPS locations of all the compounds where dog and cat fleas were obtained were recorded.
Fleas were identified using the entomological keys (Soulsby 1982) and pooled by species, sex, and host. After identification, these fleas were immediately placed in dry ice until delivery to the laboratory at KEMRI/CDC, Kisumu, Kenya, where they were stored at −80°C. A quantitative real-time PCR (qPCR) assay that amplifies and detects a 74-basepair (bp) segment of the citrate synthase (gltA) gene (Stenos et al. 2005) was used to screen nucleic acid preparations from 10% of systematically selected fleas (every 10th flea) for the presence of rickettsial DNA. Rickettsial DNA from the fleas was further tested using a species-specific Rasemb qPCR assay (Jiang et al. 2013) that amplifies and detects a 112-bp fragment of the outer membrane protein B gene (ompB). The remainder of the fleas were shipped (on dry ice) to the Naval Medical Research Center (NMRC), Silver Spring, MD, for further characterization. The collection of specimens from dogs and cats was approved by the KEMRI Ethical Review Committee and Animal Care and Use Committees (KEMRI SSC 2412).
C. canis and C. felis fleas were disinfected prior to trituration by washing once in 70% ethanol for 1 min, followed by three washes in sterile water for 1 min per wash. Each disinfected flea was triturated into 1 mL of Snyder's I buffer (0.22 M sucrose, 0.0032 M KH2PO4, 0.0086 M Na2HPO4, and 0.0049 M glutamic acid/liter, pH 7.4) using a single, sterile razor blade for each flea. Each triturated flea was divided into two aliquots; one aliquot was subjected to DNA extraction and the second aliquot was used for infection of cell lines.
Initial infections and time course infections in S2 and C6/36 cells
Drosophila S2 cells maintained in Schneider's Drosophila medium (containing 10% heat-inactivated FBS) were split into six-well tissue culture plates (60 mm) (BD Falcon, Franklin Lakes, NJ) 1 day before inoculation of the cultures with triturated fleas. At the time of inoculation, the confluence of S2 cells was 50%, and the medium (containing 10% heat-inactivated FBS) was removed from each well and replaced with medium containing 5% heat-inactivated FBS. Eight individual fleas (C. canis and C. felis) were triturated as described above. Two male and three female C. canis fleas were triturated, and one female and two male C. felis fleas were triturated. From each individual flea triturate, 900 μL of triturate was used to inoculate a single well of S2 cells and 100 μL of triturate was used for DNA extraction(s). DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, Germantown, MD) following the manufacturer's protocol for spin column DNA isolation; each triturated flea sample was eluted in 50 μL of the kit's AE elution buffer. Following inoculation, the plates were gently rocked back and forth (manually) five to six times and placed in the incubator at a temperature of 25°C. DNA was extracted from 1 mL of each inoculated culture at 1, 3, 6, 7, and 13 days postinfection (dpi). For each time period, cells and medium were removed from the culture(s), and fresh medium (5% heat-inactivated FBS) was used to replace the old medium that was removed with the cell sample. Each milliliter of cells that was removed from the wells was individually centrifuged at 11,000 rpm for 7 min; the supernatant was removed, and the cells were resuspended in 200 μL of sterile phosphate-buffered saline (PBS; Invitrogen/Life Technologies). DNA was extracted following the manufacturer's instructions for spin column DNA isolation from cultured cells (DNeasy Blood and Tissue Kit, Qiagen). Each DNA sample was subjected to our species-specific Rasemb qPCR assay (Jiang et al. 2013).
Infections were also assessed at selected time points using Acridine Orange stain (Amersham Biosciences/GE Healthcare, Piscataway, NJ). The cells were prepared on glass slides for staining by centrifuging 1 mL of cells for 7 min at 11,000 rpm (11,363 relative centrifugal force [RCF]) in an Eppendorf 5424 centrifuge, followed by resuspension of the cells in 20–50 μL of sterile PBS. Approximately 8–10 μL of the resuspended cells were then heat fixed to the slides and stained with Acridine Orange by flooding the slides with Acridine Orange for 1 min followed by rinsing in distilled water. Images were viewed with an Olympus BX43 microscope and DP72 camera (Olympus, Center Valley, PA).
At 14 dpi (following initial inoculation of the S2 cells), all five uncontaminated cultures of Candidatus R. asemboensis initiated from five different fleas (identified by sequencing of the 17-kD antigen gene, gltA, and ompB, and found to be 100% identical to each other and Candidatus R. asemboensis) were combined. The combined cultures were used to inoculate one flask of S2 cells and one flask of C6/36 cells. Each fresh flask of S2 and C6/36 cells was plated 1 day before inoculation and was at a confluency of 50% at the time of inoculation. For inoculation of the S2 cells, 2.5 mL of the combined cultures were used to inoculate a fresh flask (75 cm2) (Corning, Tewksbury, MA) of S2 cells. For inoculation of the C6/36 cells, 2.5 mL of the combined culture was disrupted mechanically by shaking with sterile, glass beads. The culture was then centrifuged at 600×g for 20 min, and all of the resulting supernatant was used to infect the C6/36 cells (75-cm2 flask) (Corning). Both flasks were gently rocked back and forth (manually) five to six times and incubated at a temperature of 25°C. DNA was prepared from the S2 and C6/36 cultures as described above at 4 and 8 dpi. At 8 dpi, each of these cultures was used to inoculate fresh cells (50% confluent); i.e., 5 mL of the S2 culture was used to inoculate fresh S2 cells and 5 mL of the C6/36 culture was used to inoculate fresh C6/36 cells. The freshly inoculated cultures were incubated at 25°C. At 45 dpi, DNA was extracted from 1 mL of each culture and/or Acridine Orange stain was applied as previously described. The 45 dpi cultures of S2 and C6/36 cells were used to inoculate (using 3 mL of infected cells) fresh cultures of S2 and C6/36 cells; the remaining cultures were frozen at −80°C using Recovery Cell Culture Freezing Medium (Invitrogen/Life Technologies). Both the C6/36 and S2 cultures infected with Candidatus R. asemboensis have been continually cultured to date as described above.
For time course infections in the C6/36 and S2 cells, the cells were split into 75-cm2 flasks (Corning) 1 day before infection with Candidatus R. asemboensis; at the time of infection cells were 50% confluent. For time course infections, 200 μL of frozen S2 or C6/36 seed material was used to infect the cells, or 1 mL of the continuous culture of Candidatus R. asemboensis was used (infected S2 cultures were used to infect fresh S2 cells and infected C6/36 cultures were used to infect fresh C6/36 cells). DNA was extracted from an 800-μL aliquot of the frozen seed material and from a 1-mL aliquot of the continuous cultures used to initiate infection in the uninfected C6/36 and S2 cells. This DNA preparation served as a representation for the number of DNA copies present at a time point of 0 dpi. At selected time points during the time course infection experiments, cells and medium were removed by drawing a pipette across the bottom of the flask(s) to remove 1 mL of the cells and medium for DNA extraction. Fresh medium was used to replace the medium that was removed with the cells for DNA each time a sample was taken from the flask(s).
qPCR for determination of infection
Candidatus R. asemboensis infections in the S2 and C6/36 cells were detected using the species-specific Rasemb qPCR assay (Jiang et al. 2013) and the Platinum qPCR UDG Supermix Kit (Invitrogen/Life Technologies, Carlsbad, CA). The species-specific RfelB qPCR assay (Odhiambo et al. 2014) and the Platinum qPCR UDG Supermix Kit (Invitrogen/Life Technologies) were used to test the S2 and C6/36 cells for the presence of R. felis. All qPCR assays were performed using a Cepheid Smart Cycler. For normalization of Candidatus R. asemboensis copy numbers, qPCR assays for the housekeeping genes Drosophila ribosomal protein 15a and A. albopictus ribosomal protein S7 were used as previously described (Schneider and Shahabuddin 2000, Luce-Fedrow et al. 2014). The relative Candidatus R. asemboensis genome levels were calculated after normalization to the housekeeping gene(s). The Candidatus R. asemboensis genome levels were calculated using a standard curve that was constructed using 10-fold dilutions of known concentrations of a Candidatus R. asemboensis ompB target sequence cloned into a plasmid. For negative controls, qPCR reactions of uninfected S2 cells, uninfected C6/36 cells, and reactions containing no DNA template were included with each qPCR run. No copies of Candidatus R. asemboensis or R. felis were detected in the negative controls.
DNA sequencing of Candidatus R. asemboensis
DNA samples extracted from Candidatus R. asemboensis–infected C6/36 and S2 cells and from uninfected C6/36 and S2 cells were used in PCR assays targeting segments of the 17-kD antigen gene, gltA, and ompB genes, as previously described (Jiang et al. 2013). All PCR and nested PCR reactions were performed using the Platinum PCR Supermix High-Fidelity Kit (Invitrogen/Life Technologies). PCR amplicons were purified using the Qiaquick PCR Purification Kit (Qiagen), and DNA sequencing reactions were completed using the ABI PRISM 3.1 BigDye Terminator Cycle Sequencing Kit (Applied BioSystems, Foster City, CA). Sequencing products were purified using Performa DTR Gel Filtration Cartridges (Edge Bio, Gaithersburg, MD) and were sequenced on an automated ABI Prism 3500 Genetic Analyzer (Applied Biosystems). Geneious Pro version 5.5.7 was used to complete the sequence analysis (Geneious Pro 5.5.7 2014).
Transmission electron microscopy
Infected (14 dpi) C6/36 cells and uninfected C6/36 cells were prepared for transmission electron microscopy (TEM). Cells were removed from the flask(s) by drawing a pipette across the bottom of the flask(s) to remove the cells/media, which were centrifuged for 5 min at 1200 rpm. The supernatants were discarded, and the samples were immersion fixed in a fixative containing 4% formaldehyde/1% glutaraldehyde in 0.1 M sodium phosphate buffer. The samples were fixed for 1 h, washed three times with 0.1 M sodium phosphate buffer (15 min/wash), and then postfixed with 1% osmium tetroxide I and 0.1 M sodium phosphate buffer for 1 h. The images were collected on a JEM 100 CX II transmission electron microscope (JEOL, Japan).
Results
Confirmation of Candidatus R. asemboensis–positive fleas
Over 700 fleas were collected from 44 dogs, among which were C. felis and C. canis fleas. Screening of approximately 10% (n=70) of the fleas with a gltA genus–specific (Stenos et al. 2005) and Rasemb species-specific (Jiang et al. 2013) qPCR assay revealed that >98% of the fleas were positive for Candidatus R. asemboensis, with Ct values ranging from 22.43–36.27.
Isolation of Candidatus R. asemboensis into S2 and C6/36 cells lines
To isolate Candidatus R. asemboensis in cell culture, we used eight individual C. canis or C. felis fleas to inoculate Drosophila S2 cells. One day following inoculation, the cultures were assessed visually for evidence of contamination; three of the cultures appeared contaminated and were discarded (thus eliminated from the isolation procedure). Evidence of infection/identification of rickettsiae in the cells was determined by using cytological staining and molecular techniques. The Rasemb qPCR assay was used to confirm that the triturated fleas used for initial inoculation of the S2 cells were infected with Candidatus R. asemboensis and to monitor the infection in the S2 cells at 1, 3, 6, 7, and 13 days following the initial inoculation. The five fleas used for the initial inoculation of the S2 cells were assessed by the Rasemb qPCR assay and found to contain a total of 119,364–1,566,911 DNA copies (genome equivalents) of Candidatus R. asemboensis per flea, with an average of 737,566 DNA copies of Candidatus R. asemboensis per flea.
A volume of 900 μL of flea triturate was used to inoculate each culture of S2 cells; thus, the initial inoculums contained a range from 107,427 to 1,157,492 DNA copies of Candidatus R. asemboensis (Fig. 1, time point 0 dpi). An overall decrease in the number of Candidatus R. asemboensis DNA copies per milliliter of cells sampled from each culture was observed for all of the five inoculated cultures when comparing the initial inoculum for each culture to the number of rickettsial DNA copies present at 13 dpi (Fig. 1). At 13 dpi, we observed as few as 2990 Candidatus R. asemboensis DNA copies per millimeter of cell culture in culture S2-6 and as many as 65,505 copies per milliliter of cell culture in culture S2-1 (Fig. 1).
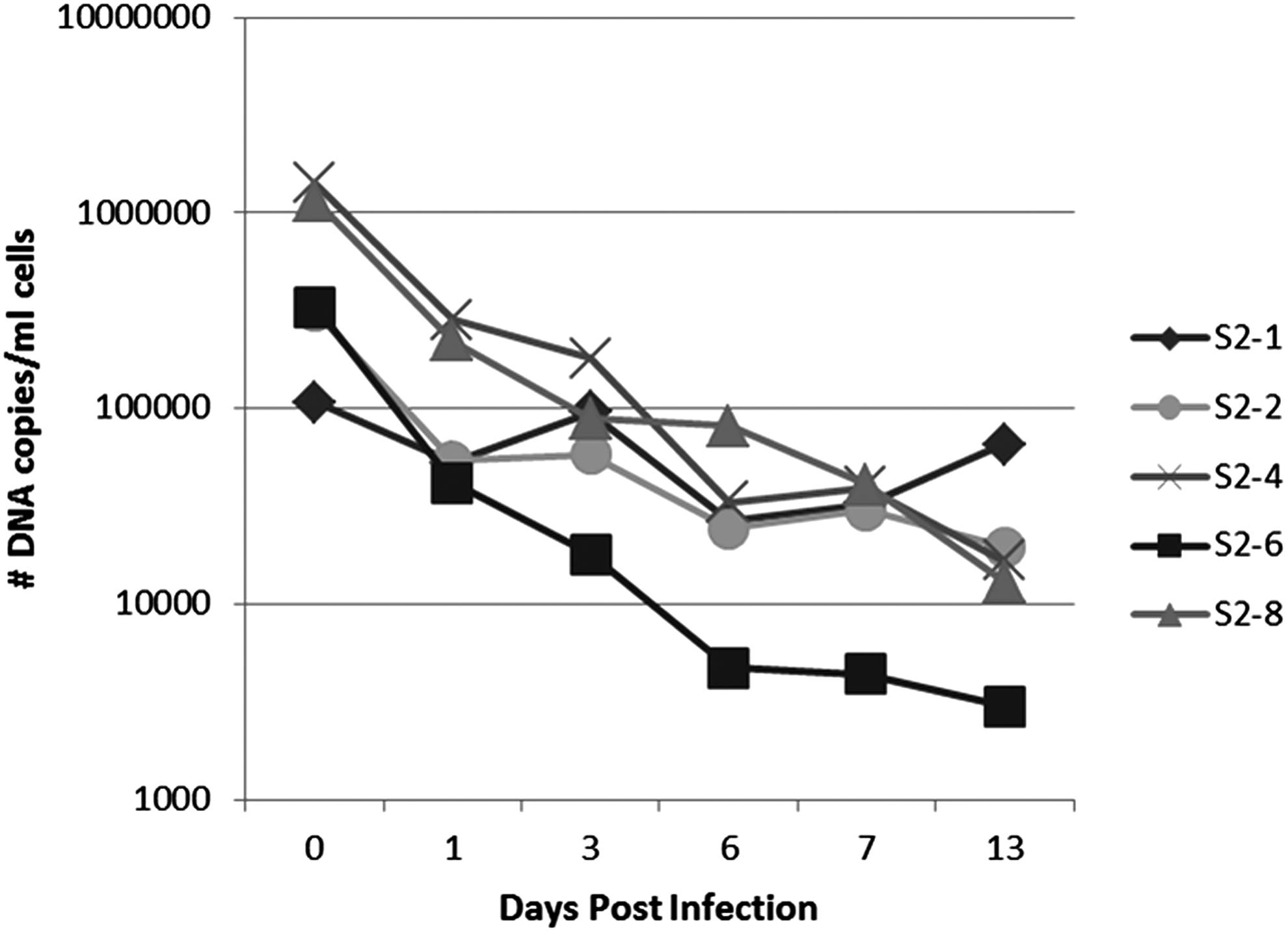
Initial isolation of Candidatus R. asemboensis in Drosophila melanogaster S2 cells. The five initial S2 cultures (S2-1, S2-2, S2-4, S2-6, S2-8) each inoculated with a single flea triturate (fleas 1, 2, 4, 6, or 8) are shown. The numbers of DNA copies present at the indicated time points were quantitated using a Candidatus R. asemboensis–specific qPCR assay. No rickettsial copies were detected in uninfected cells (data not shown).
With the observation that the number of Candidatus R. asemboensis DNA copies was either declining or slightly increasing (culture S2-1), we combined all of the cultures and used the combination to inoculate a single flask of S2 cells and a single flask of C6/36 cells. These cultures were also monitored for presence of Candidatus R. asemboensis using our Rasemb qPCR assay; samples were taken from each flask at 4 and 8 dpi. During this second passage of Candidatus R. asemboensis, we observed an initial decrease in the number of rickettsial DNA copies from the time of initial inoculation (0 dpi) until 4 dpi, but then observed a subsequent increase in the number of copies from 4 dpi to 8 dpi in both the S2 and C6/36 cultures (Fig. 2).
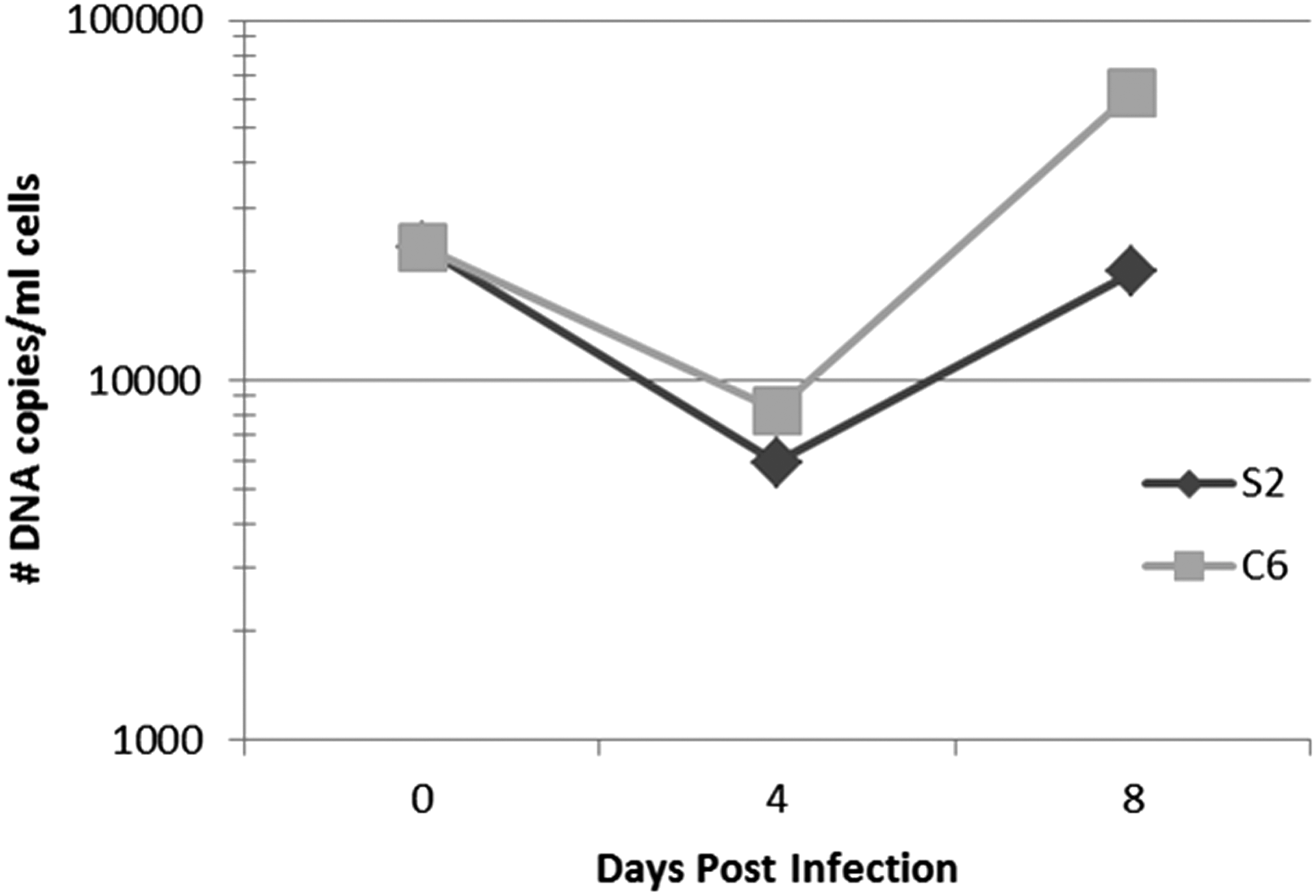
Passage number 2 of Candidatus R. asemboensis in Drosophila melanogaster S2 cells or A. albopictus C6/36 cells. The number of DNA copies present at the indicated time points were quantitated using a Candidatus R. asemboensis–specific qPCR assay. Rickettsial DNA was not detected in uninfected cells (data not shown).
Following this observation, we used the 8 dpi, second-passage cultures to initiate a third passage of the bacteria in one fresh flask of S2 cells and one of C6/36 cells. These cultures were incubated undisturbed for 45 days. Upon Acridine Orange staining at 45 dpi, Candidatus R. asemboensis could be observed in both the infected S2 and C6/36 cultures; no rickettsiae were seen in uninfected cells (Fig. 3). After this third passage of the Candidatus R. asemboensis in the S2 and C6/36 cells, the cultures have been maintained in both cells lines and have also been frozen at −80°C for use as future seed material. To date, the bacteria have been passaged in each cell line 12 times, with the passaging occurring at 18 dpi to as long as 68 dpi and with the average number of days between passages being 40 days. For one set of cultures, we continuously maintained the infection for upward of 100 days by periodically removing a portion of the spent medium (approximately half), adding new medium (the same amount that was removed), and adding uninfected cells every 15–30 days. During this lengthy time course, rickettsiae could be observed by Acridine Orange stain in large numbers both extra- and intracellularly, often occurring in pairs and/or chains (Fig. 4). This indicated that continuous culture of the bacteria could be performed well beyond the average passage time of 40 days.

Acridine Orange staining of Candidatus R. asemboensis in Drosophila melanogaster S2 cells (
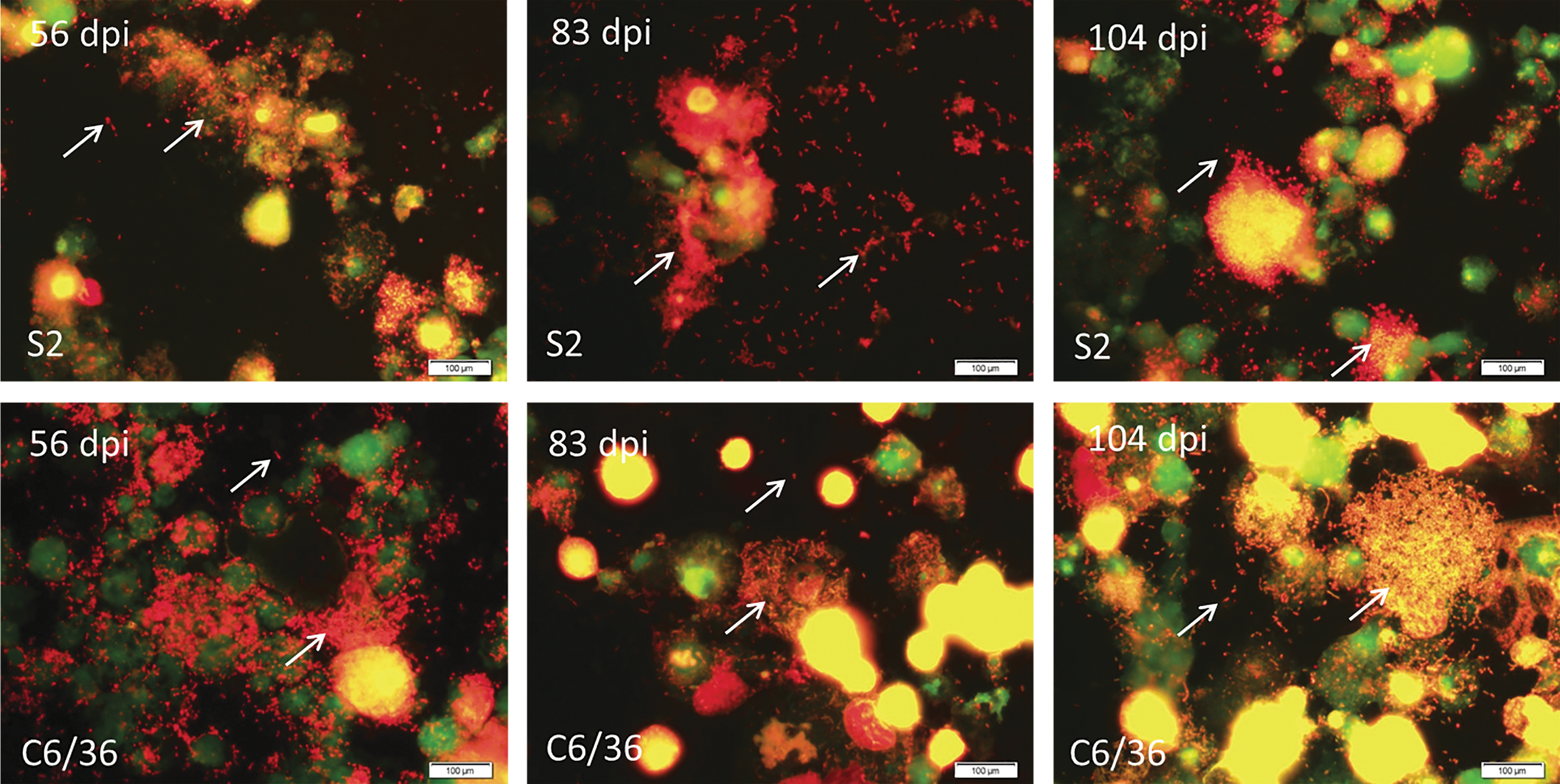
Acridine Orange staining of Candidatus R. asemboensis Drosophila melanogaster S2 and Aedes albopictus C6/36 cells at various time points. Rickettsiae were not observed in uninfected cells (negative, not shown). Images were captured using an Olympus BX43 microscope and DP72 camera with a 10×ocular and 100×objective. Bars, 100 μm.
To compare the kinetics of Candidatus R. asemboensis infection in the S2 and C6/36 cells lines, we employed three independent time course infection studies in each cell line. For the first time course infection (S2-1 and C6/36-1), we used frozen seeds of Candidatus R. asemboensis to initiate the infections; for the second and third time courses (S2-2, S2-3 and C6/36-2, C6/36-3), we used seeds of Candidatus R. asemboensis that were currently in continuous culture. Rickettsial DNA was detected at each time point for all experiments in both the S2 and C6/36 cells (Fig. 5). In the S2 cells, we observed an initial increase in the average number of Candidatus R. asemboensis copy numbers during the time range of 3–10 dpi (between 7–10 dpi for S2-1 and between 0–3 dpi for S2-2 and S2-3) followed by a subsequent decrease in average copy numbers to 31 dpi (Fig. 5B). However, at 31 dpi there were more average bacterial copies present for S2-1, S2-2, and S2-3 than were present in the initial dose; e.g., approximately 700 more copies at 31 dpi for S2-1, 5.6×105 more copies for S2-2, and 7.7×104 for S2-3 (Fig. 5B). In the C6/36 cells, we observed an initial decline from 0 to 5 dpi in the average number of copies of Candidatus R. asemboensis (between 3 and 5 dpi for C6/36-1 and between 0 and 3 dpi for C6/36-2 and C6/36-3) (Fig. 5A). After 5 dpi, we observed a continual increase in the average number of Candidatus R. asemboensis copies in C3/36-1, C6/36-2, and C6/36-3 time courses (Fig. 5A). By 31 dpi there were an average of 5.1×108, 1.9×1010, and 9.7×109 Candidatus R. asemboensis copies/mL of cells sampled in time courses C6/36-1, C6/36-2, and C6/36-3, respectively, which were all increased compared to the inoculating dose (0 dpi) (Fig. 5A).
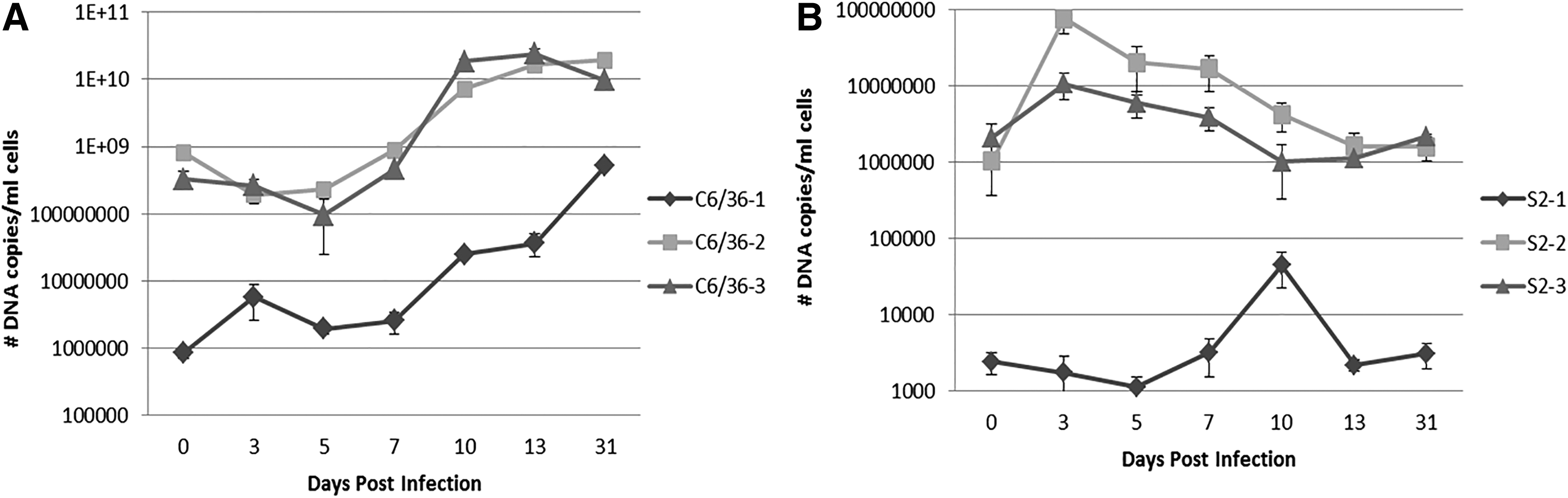
Comparison of growth of Candidatus R. asemboensis in A. albopictus C6/36 cells (
These data suggest that Candidatus R. asemboensis is capable of replicating/completing its life cycle in both the S2 and C6/36 cells. When comparing the average number of rickettsiae present in the S2 versus the C6/36 cells during the time course infections, the C6/36 cells contained more bacteria. We acknowledge that the C6/36 time courses were initiated with larger doses of bacteria. However, when comparing time courses C6/36-1 to S2-2, which were initiated with similar quantities of bacteria (approximately 1 log more in the C6/36-1 inoculum), the Candidatus R. asemboensis appears to replicate more rapidly in the C6/36 cells.
In addition to using our species-specific Rasemb qPCR assay (Jiang et al. 2013) as a tool to determine that our isolate was indeed Candidatus R. asemboensis, we also used the species-specific RfelB assay (Odhiambo et al. 2014) to confirm the absence of R. felis in our cultures. The time course samples were all positive for Candidatus R. asemboensis and negative for R. felis (data not shown).
Transmission electron microscopy
To visualize Candidatus R. asemboensis within the cells, TEM was performed on C6/36 cells that were infected for 14 days or on uninfected C6/36 cells. Several cells were observed in the TEM preparations that contained numerous rickettsiae. Round forms of rickettsiae were primarily observed in addition to a few rickettsiae of slightly elongated shape in the cytoplasm of the infected cells; no rickettsiae could be seen in the nuclei of the infected cells (Fig. 6). The round forms of rickettsiae ranged in diameter from approximately 0.375 to 0.5 μm; the slightly elongated forms of rickettsiae were approximately 0.5–0.625 μm in length and approximately 0.25–0.375 μm in width/diameter. The rickettsial slime layer(s), as evidenced by an electron-lucent “halo” surrounding the rickettsiae within cytoplasm of the cells, could be observed at low and higher magnifications (Fig. 6). At higher magnifications, the presence of a cell wall membrane as well as a cytoplasmic membrane, with a defined periplasmic space between them, could be discerned for individual rickettsiae. Our current TEM analysis does not provide evidence that Candidatus R. asemboensis grows within the nucleus of the host cell. However, additional analysis of samples prepared from time points beyond 14 dpi and samples grown in additional host cells will be necessary to make a definitive conclusion.
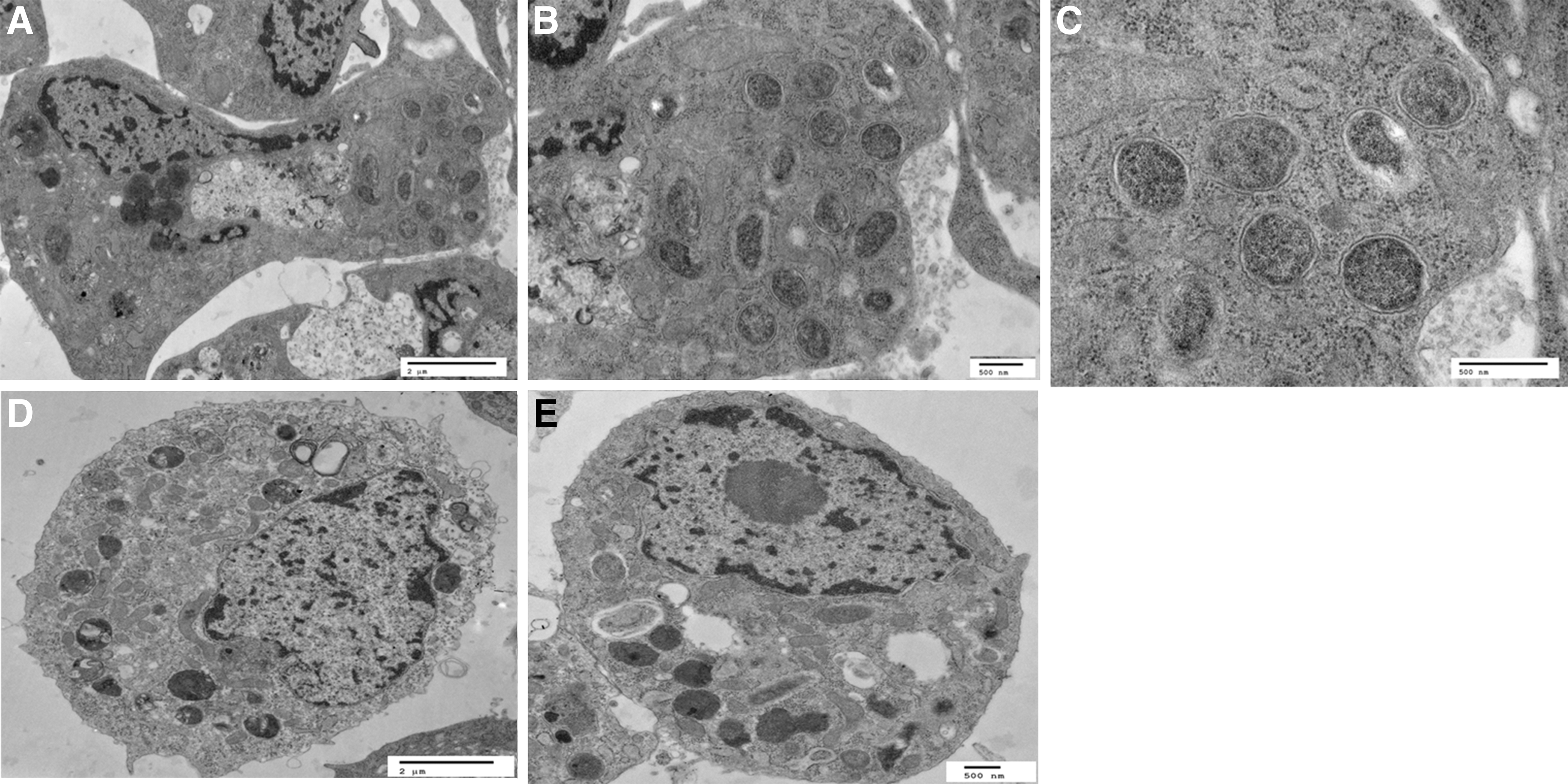
Transmission electron micrographs of Candidatus R. asemboensis in A.albopictus C6/36 cells at 14 days postinfection (dpi) (the same cell is represented in images
DNA sequencing confirms the isolation of Candidatus R. asemboensis
DNA samples that were extracted from Candidatus R. asemboensis–infected C6/36 and S2 cells were amplified by PCR reactions that targeted fragments of the 17-kD antigen gene (389 bp), gltA (1186 bp), and ompB (536 bp) genes, as previously described (Jiang et al. 2013). The amplified fragments of the 17-kD antigen gene displayed 100% sequence identity to several corresponding 17-kD antigen gene fragments, including: Rickettsia species F30 (JN315969); Rickettsia species F82 (JN315975) (both Candidatus R. asemboensis molecular isolates); C. felis isolate F144 (JF511462); Rickettsia species SE313 (DQ166937); and Rickettsia endosymbiont of C. felis F143 (JF511461) (Table 1). The amplified gltA gene fragments from our isolates all displayed 100% similarity to the corresponding gltA fragments of Rickettsia species F30 (JN315968) and Rickettsia species F82 (JN315974) (Table 1). The next closest corresponding matches to the amplified gltA fragment included a 99.6% match to Rickettsia species RF2125 (AF516333), a 98.3% match to an uncultured Rickettsia species clone ric_Ag_101730 (JN620082), and a 98% match to Rickettsia species SG101 (GQ255903) (Table 1). For the amplified fragments of the ompB gene, all samples displayed a 100% match to the corresponding ompB fragment of Rickettsia species F30 (JN315972) (Table 1). The next closest matches to the corresponding ompB fragment included a 93% match to Rickettsia species 518 clone 1 (EU430242), a 93% match to R. felis clone Ar3 (GQ385243), and a 93% match to R. felis URRWX Cal 2 (CP000053) (Table 1). The results for DNA sequencing of the 17-kD antigen gene, gltA, and ompB genes confirmed the identity of the Candidatus R. asemboensis isolates and that isolation into the S2 and C6/36 cell lines was successful.
The closest validated Rickettsia sp. for 17-kD antigen gene, gltA, and ompB sequences is R. felis.
Discussion
In this study, we have described the isolation and cultivation of Candidatus R. asemboensis from C. canis and C. felis fleas collected in Asembo region of western Kenya (Jiang et al. 2013). We demonstrated the isolation and growth of the bacteria in Drosophila S2 and A. albopictus C6/36 cells lines using molecular assays and cytological tools for visualization of the bacteria. During our time course infections of S2 and C6/36 cells with Candidatus R. asemboensis, we observed a difference in the growth kinetics of the bacteria in each cell line during the 31-day periods of the experiments. In the C6/36 cells, we observed a proposed lag phase from approximately 0 to 7 dpi followed by an exponential phase from 7 to 31 dpi. These kinetics are similar to those observed when propagating R. felis strain LSU in C6/36 cells (Luce-Fedrow et al. 2014). In contrast, the lag phase of Candidatus R. asemboensis in the S2 cells appears to last much longer, e.g., for the 31-day duration of the time course. However, at 31 dpi, we could detect an increase in the number of molecular copies of Candidatus R. asemboensis compared to the inoculating dose, suggesting that the bacteria were replicating and hence the time frame surrounding 31 dpi may be the start of the exponential phase of growth in the S2 cells.
Moreover, we have observed large numbers of bacterial copies present in our continuously infected cultures of S2 cells at 35, 40, and 68 dpi (4.2×108, 1.1×108, and 8.8×107 bacterial copies/mL of cells, respectively) (data not shown). This may indicate that growth of Candidatus R. asemboensis is much slower in the S2 cells, with a longer lag phase followed by a later exponential phase, as compared to the C6/36 cells. Differences in growth kinetics of rickettsiae among cell lines have been observed previously. Rickettsia raoultii was found to be pathogenic for Rhipicephalus (Boophilus) microplus and Dermacentor nitens cell lines, but exhibited little growth in Dermacentor albopictus and Dermacentor andersoni cells lines (Alberdi et al. 2012). Similarly, Rickettsia peacockii was found to be pathogenic for Rhipicephalus (B.) microplus cells but demonstrated poor growth in Dermacentor cells lines (Kurtti et al. 2005). Thereby, the differences in rickettsial growth between our insect cell lines are not atypical, and we hypothesize that the differences may be attributable to specific host factors that have yet to be deduced.
Acridine Orange staining and TEM were used to detect Candidatus R. asemboensis within the host cells. No marked cytopathic/cytotoxic changes to cells infected with Candidatus R. asemboensis were observed throughout our continuous culture of the bacteria and/or during our time course infections. We have cultured the bacteria for upward of 70 days by adding additional medium to the flasks; moreover, we have kept some cultures of the bacteria in culture for over 100 days by removing and replacing spent medium and adding fresh cells to the culture. Even during these long periods of infection, the only notable cytopathic event was a lysis of cells that became overly parasitized by the bacteria (as observed by Acridine Orange staining). Intracellular invasion of the cells following infection with Candidatus R. asemboensis could be easily observed by Acridine Orange staining as early as 3 dpi. Before this time point, most of the bacteria appeared to remain extracellularly. In the C6/36 cells, we observed heavily parasitized cells as early as 14 dpi; in the S2 cells, heavy parasitization of the cells occurred at approximately 25–30 dpi. Bacteria usually appeared in singlets and/or doublets as observed by Acridine Orange staining; we observed very few instances of the bacteria forming long chains either intra- or extracellularly. We were unable to confirm the intranuclear growth of Candidatus R. asemboensis using Acridine Orange stain. We observed a few instances in which bacteria appeared to be within the nucleus of the cell(s); however, this could not be confirmed using TEM, and thus more experiments will be necessary to establish whether or not Candidatus R. asemboensis grows intranuclearly in S2 and/or C6/36 cells.
TEM analysis performed on C6/36 cells that were infected with Candidatus R. asemboensis for 14 days revealed multiple cells containing free rickettsiae in the cytoplasm. The majority of the intracellular rickettsiae were round in shape, of normal rickettsial size, and observed primarily in the cytosol of the cells. We also observed a few rickettsiae of slightly elongated shape present in the cytosol of the cells, but none that exceeded the normal size of rickettsiae, as has been previously observed for other rickettsial species (Wisseman and Waddell 1975, Labruna et al. 2004, Sunyakumthorn et al. 2008). Additional studies using alternate host cells, variations in growth temperature/time, and/or changes in cell culture media will be necessary to determine if Candidatus R. asemboensis develops a long-form phenotype.
To confirm that our rickettsial isolate growing in the S2 and C6/36 cells was Candidatus R. asemboensis, we used DNA sequencing analysis. Partial sequences of the 17-kD antigen gene, gltA, and ompB genes amplified from our culture isolates were sequenced and examined for homology to the corresponding nucleotide sequences available in GenBank. When the 17-kD antigen gene of Candidatus R. asemboensis was compared to known sequences, it was found to have 100% identity to Rickettsia species isolates F30 and F82, which are molecular isolates of Candidatus R. asemboensis, and to Rickettsia species isolates F144, SE313, and F143. Rickettsia species isolates F30 and F82 were first identified in fleas collected from Asembo, Kenya, and proposed to be considered a new rickettsial species, Candidatus R. asemboensis (Jiang et al. 2013).
Rickettsia species F144 and F143 were first described in fleas collected from dogs in Bangkok, Thailand (Foongladda et al. 2011), and Rickettsia species SE313 was first described in fleas and mites collected from rats in Egypt (Loftis et al. 2006). When comparing the sequence of the gltA gene from our isolates, we also found a 100% match to Rickettsia species F30 and F82. Beyond F30 and F82, the pathogens closest in similarity to the gltA sequence obtained from our isolate included Rickettsia species RF2125, Rickettsia species clone ric_Ag_101731, and Rickettsia species SG101, all R. felis–like organisms. RF2125 has been described in fleas collected from three different continents, e.g., from the Thai–Myanmar border (Parola et al. 2003), the United States (Reeves et al. 2005, Nelder et al. 2009), and Hungary (Hornok et al. 2010). Rickettsia species clone ric_Ag_101731 was first reported in Anopheles gambiae mosquitoes in Cote d'Ivoire (Socolovschi et al. 2012), and Rickettsia species SG101 was first described in tsetse flies in Senegal (Mediannikov et al. 2012). The partial sequence of the ompB gene amplified from our isolate also demonstrated a 100% match to Rickettsia species F30 and F82. The next closest corresponding matches to the ompB fragment of our isolate included a 93% match to each of the following: Rickettsia species 518 clone 1, which was detected in Ixodes tasmani ticks collected in Australia (Vilcins et al. 2009); R. felis clone Ar3, which was identified in Liposcelis bostrychophila collected in Australia and the United States; and to R. felis URRWX Cal2. The high similarity of our isolate to the aforementioned isolates suggests that it can be considered a R. felis–like organism. The cosmopolitan nature of R. felis–like organisms, as demonstrated by their prevalence in a variety of potential vectors across the world, gives reason for concern about their potential public health impact if they are indeed confirmed as human pathogens. Isolation of these types of agents in cell culture represents a step toward being able to better define their pathogenicity in mammalian hosts.
Consequently, we have demonstrated that Candidatus R. asemboensis can be isolated from Ctenocephalides fleas and propagated in cell culture. Whole-genome sequencing of this new organism has been completed by our laboratory, the results of which have been submitted for publication (Jima et al. 2015), and will be useful for contributing to a better understanding of the genetic features of this newly isolated organism. Moreover, the isolation of Candidatus R. asemboensis in pure culture represents a milestone toward validation of the organism as a new species and for the onset of experiments for determining its potential virulence, which we hope will ultimately be useful in providing additional insight translatable to other R. felis–like organisms.
Conclusions
Candidatus Rickettsia asemboensis is a newly described rickettsial species, closely related to R. felis, which causes flea-borne spotted fever in humans. Candidatus R. asemboensis was first molecularly described in fleas collected from Asembo, Kenya, and it is not known whether the bacteria are pathogenic to humans. We have described the first in vitro isolation and growth of Candidatus R. asemboensis from dog and cat fleas collected in Kenya. Molecular and cytological techniques were used to characterize and visualize our isolate. We used molecular methods (qPCR) to detect the bacteria in vitro and Acridine Orange staining to visualize the bacteria in the host cells. DNA sequencing of several genes indicates that our isolate is similar to several other R. felis–like organisms. This is the first report of isolation and maintenance of a R. felis–like organism in vitro.
Footnotes
Acknowledgments
We thank the IHAP team (Samuel Asembo, Michael Otieno, James Oigo, Pauline Otieno, and Julius Ouma) for their assistance during the collection of flea specimens from dogs and cats. This work is supported by the Global Emerging Infections Surveillance and Response System, a Division of the Armed Forces Health Surveillance Center, work unit number 0000188M.0931.001.A0074.
The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, nor the US Government.
Author Disclosure Statement
I am an employee of the US Government (A.L.R.) and this work was prepared as part of my official duties. Title 17 U.S.C. §105 provides that “Copyright protection under this title is not available for any work of the United States Government.” Title 17 U.S.C. §101 defines a US Government work as a work prepared by a military service member or employee of the US Government as part of that person's official duties.