
Abstract
Astroviruses are a major cause of gastroenteritis in humans and animals. Recently, novel groups of astroviruses were identified in apparently healthy insectivorous bats. We report the detection of diverse novel astrovirus sequences in nine different European bat species: Eptesicus serotinus, Hypsugo savii, Myotis emarginatus, M. mystacinus, Nyctalus noctula, Pipistrellus nathusii or P. pygmaeus, P. pipistrellus, Vespertilio murinus, and Rhinolophus hipposideros. In six bat species, astrovirus sequences were detected for the first time. One astrovirus strain detected in R. hipposideros clustered phylogenetically with Chinese astrovirus strains originating from bats of the families Rhinolophidae and Hipposideridae. All other Czech astrovirus sequences from vesper bats formed, together with one Hungarian sequence, a separate monophyletic lineage within the bat astrovirus group. These findings provide new insights into the molecular epidemiology, ecology, and prevalence of astroviruses in European bat populations.
Introduction
B
In the present study, we investigated the occurrence and genetic diversity of AstVs in various species of bats in the Czech Republic. We compared the partial genome sequences of detected AstV strains with published sequences and then conducted a phylogenetic analysis.
Materials and Methods
A total of 43 fecal or intestinal content samples from different European bat species were collected in 2008-2014 in South Moravia, Czech Republic (Table S1) (Supplementary Data are available at
Total RNA was extracted with the QIAamp Viral RNA Mini Kit (Qiagen) from the supernatant of 10% (vol/vol) fecal or intestinal content suspensions in phosphate-buffered saline (PBS; 0.1 M, pH 7.2) following the manufacturer's protocol. Reverse transcription was carried out in a 20-μL volume containing 150 ng of random hexamers using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Mannheim, Germany) according to the manufacturer's recommendations. Random hexamer-generated cDNA was screened for the presence of AstV nucleic acid by seminested PCR that targeted the most conserved region of the RNA-dependent RNA polymerase (RdRp) gene (Chu et al. 2008). Resulting amplicons were purified with the Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI) and sequenced commercially (SEQme, Dobříš, Czech Republic).
To determine whether the mixed fecal sample from two bat species contained diverse AstV strains, the PCR product was cloned into pCR2.1-TOPO plasmids (Invitrogen, Germany). Multiple clones were picked, purified with a PureLink Quick Plasmid Miniprep Kit (Invitrogen, Germany), and sequenced with M13 forward and reverse primers. The RdRp nucleotide dataset was translated into amino acids and aligned using the ClustalW algorithm in BioEdit 7.0.5.3 with default parameters (Hall 1999). The edited nucleotide dataset was used for phylogenetic analysis. Bayesian inference tree was computed with MrBayes 3.1.2 (Huelsenbeck and Ronquist 2005), with five million generations under the general time reversible (GTR)+I+γ model with gamma distribution in six categories. Consensual topology was created based on 45,000 trees.
Results
Out of 43 samples from 15 different bat species, nine samples (20.9 %) from nine species (E. serotinus, H. savii, M. emarginatus, M. mystacinus, N. noctula, P. nathusii or pygmaeus, P. pipistrellus, V. murinus, and R. hipposideros) tested positive for astrovirus RNA (Table S1). For the mixed sample (P. nathusii and P. pygmaeus), we could not determine which of the two species harbored the virus. Sequencing results of multiple clones of the PCR product confirmed that the mixed sample contained only one AstV strain.
Phylogenetic analysis based on partial (336 nucleotides) RdRp gene sequences clustered the newly described AstV sequences together with other bat AstV strains within the genus Mamastrovirus (Fig. 1). Of the nine AstV sequences detected in this study, seven were selected for phylogenetic analysis. Two sequences were not suitable for phylogeny because of insufficient length. All but one of the AstV sequences grouped (with high posterior probability) together in a single monophyletic lineage, along with the Hungarian sequence BatAstV/BS50/HUN/2013. The sequence detected in R. hipposideros was the most diverse and clustered with two Chinese strains originating from bats of the families Rhinolophidae and Hipposideridae (Zhu et al. 2009) (Fig. 1). The Czech bat AstV sequences showed 68.7–99.3% amino acid identities to one another. The genome sequences of AstV strains described in this study were deposited in GenBank under accession numbers KP843558–KP843564.
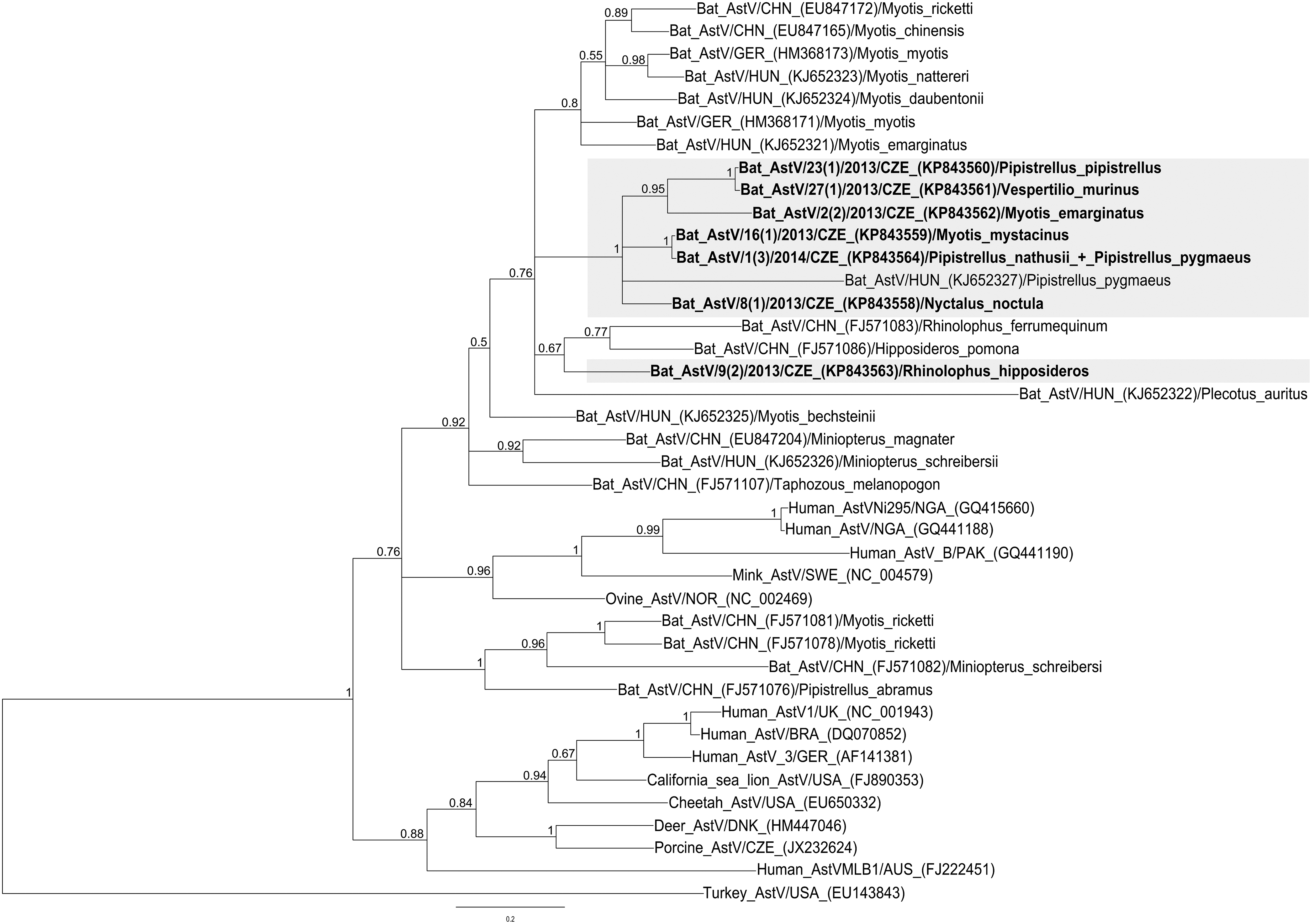
Bayesian phylogenetic tree based on a 336-nucleotide fragment within the RNA-dependent RNA polymerase (RdRp) gene of selected astroviruses. Sequence Turkey_AstV/USA (EU143843) was used as the outgroup. Numbers at nodes indicate Bayesian posterior probability. Astrovirus sequences determined in this study are highlighted and in bold.
Discussion
In the present study, we detected AstV sequences in nine out of 15 investigated bat species from South Moravia, and in six of them for the first time: E. serotinus, H. savii, M. mystacinus, P. pipistrellus, V. murinus, and R. hipposideros. Some species (e.g., E. serotinus, M. emarginatus, M. mystacinus, and R. hipposideros) harbored AstV sequences, even though their sample number was small. These findings are consistent with previous studies, which showed that many species of insectivorous bats can carry astroviruses (Chu et al. 2008, Kemenesi et al. 2014b). Interestingly, AstV sequences originating from different bat species of the family Vespertilionidae clustered together and shared higher rates of sequence identities than with AstV sequences derived from the same bat species but from different geographic areas. This observation is supported by a partial result: i.e., the sequence detected in M. mystacinus and sequence identified in the mixed fecal sample from P. nathusii/pygmaeus, shared a high level of amino acid sequence identity (99.3%) and likely represent the same AstV strain circulating in different species of bats. However, the only Czech AstV sequence demonstrated in a bat of the family Rhinolophidae (horseshoe bats) clustered differently; this sequence clustered with AstV sequences of Chinese bats from the same family and a related family. According to our results and previous reports (Zhu et al. 2009, Xiao et al. 2011), there is no significant phylogenetic clustering of bat AstVs among the different bat species and geographical regions. Viruses isolated from a single bat species at a single habitat could be clustered into different groups. On the other hand, viruses detected in different habitats could be clustered within a single group (Xiao et al. 2011).
Footnotes
Acknowledgments
This study was funded by grant no. LO1218 of the MEYS of the Czech Republic under the NPU I program. Computational resources were provided by MetaCentrum under the program LM2010005 and by CERIT-SC under the program Centre CERIT Scientific Cloud, part of the Operational Program Research and Development for Innovations (reg. no. CZ.1.05/3.2.00/08.0144). T.C. was supported by the project Postdok_BIOGLOBE—CZ.1.07/2.3.00/30.0032, co-financed by the European Social Fund and the state budget of the Czech Republic.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.