
Abstract
Although based on very limited M and L segment sequences, Artybash virus (ARTV) was proposed previously as a unique hantavirus harbored by the Laxmann's shrew (Sorex caecutiens). To verify this conjecture, lung tissues from 68 Laxmann's shrews, captured during 2006 to 2014 in eastern Siberia, Russia, and Hokkaido, Japan, were analyzed for ARTV RNA using reverse transcription polymerase chain reaction (RT-PCR). ARTV RNA was detected in six Laxmann's shrews. Pairwise alignment and comparison of partial- and full-length S, M, and L segment sequences from these Laxmann's shrews, as well as phylogenetic analyses, using maximum likelihood and Bayesian methods indicated that ARTV was distinct from other soricine shrew-borne hantaviruses and representative hantaviruses harbored by rodents, moles, and bats. Taxonomic identity of the ARTV-infected Laxmann's shrews was confirmed by full-length cytochrome b mitochondrial DNA sequence analysis. Our data indicate that the hantavirus previously known as Amga virus (MGAV) represents genetic variants of ARTV. Thus, the previously proposed designation of ARTV/MGAV should be replaced by ARTV.
Introduction
R
Seewis virus (SWSV), originally detected in the Eurasian common shrew (Sorex araneus) in Switzerland (Song et al. 2007), has now been found across the vast distribution of its soricine reservoir host in the Czech Republic (Schlegel et al. 2012), Finland (Kang et al. 2009a, Ling et al. 2014), Germany (Schlegel et al. 2012), Hungary (Kang et al. 2009a), Poland (Gu et al. 2014), Russia (Yashina et al. 2010), Slovakia (Schlegel et al. 2012), and Slovenia (Korva et al. 2013; Resman et al. 2013).
Genetically distinct hantaviruses have also been detected in other Sorex species, including Ash River virus in the North American masked shrew (Sorex cinereus) and Jemez Springs virus in the dusky shrew (Sorex monticolus) (Arai et al. 2008a), Kenkeme virus in the Asian flat-skulled shrew (Sorex roboratus) (Kang et al. 2010), Asikkala virus in the Eurasian pygmy shrew (Sorex minutus) (Radosa et al. 2013), Yakeshi virus in the taiga shrew (Sorex isodon) (Guo et al. 2013), Qian Hu Shan virus in the stripe-back shrew (Sorex cylindricauda) (Zuo et al. 2014), and Sarufutsu virus in the long-clawed shrew (Sorex unguiculatus) (Arai et al. unpublished data).
In addition, based on very limited M and L segment sequences detected in a Laxmann's shrew (Sorex caecutiens) captured near Teletskoye Lake in the Altai Republic of western Siberia, a new hantavirus, named Artybash virus (ARTV), has been proposed. In an attempt to validate its reservoir host species and explore the genetic diversity of ARTV, we analyzed lung tissues collected from Laxmann's shrews trapped in eastern Siberia, Russia, and Hokkaido, Japan. Genetic and phylogenetic analyses, based on partial- and full-length genomes of a hantavirus formerly called Amga virus (MGAV), show that it represents genetic variants of ARTV. Thus, instead of referring to this hantavirus as ARTV/MGAV, as previously proposed (Bennett et al. 2014), ARTV should be the preferred designation.
Materials and Methods
Trapping and sample collection
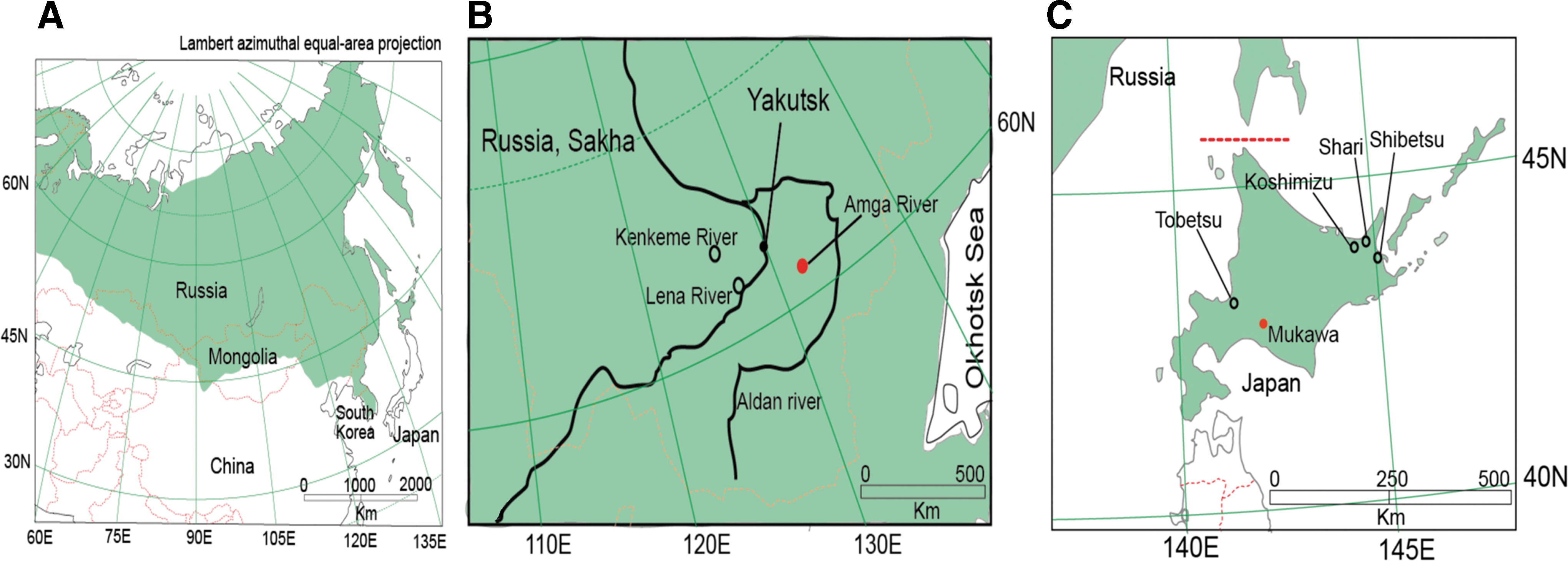
Laxmann's shrews, which are widely distributed from northern Fennoscandia through Siberia and across northern Japan (Fig. 1A), were captured at three sites in the Sakha Republic, Russia, during July and August 2006 (Fig. 1B and Table 1) and five sites in Hokkaido, Japan, between 2008 and 2014 (Fig. 1C and Table 1). Protocols for trapping, euthanasia, and tissue processing were performed according to well-established guidelines (Animal Care and Use Committee 1998).
RNA extraction and reverse transcription polymerase chain reaction
Total RNA was extracted from lung tissues using the PureLink Micro-to-Midi Total RNA Purification Kit (Invitrogen, San Diego, CA) or MagDEA® RNA 100 Kit (Precision System Science; PSS, Matstudo, Japan). cDNA was synthesized using SuperScript III First-Strand Synthesis System (Invitrogen) or PrimeScript™ II 1st strand cDNA Synthesis Kit (Takara Bio, Otsu, Japan) and an oligonucleotide primer (OSM55: 5′-TAGTAGTAGACTCC-3′) designed from the genus-specific conserved 3′-end of the S, M, and L segments of all hantaviruses. For initial screening by RT-PCR, primers were based on highly conserved regions of shrew-borne hantavirus genomes: S (outer: OSM55F, HTN-S6: 5′-AGCTCNGGATCCATNTCATC-3′; inner: Cro2F: 5′-AGYCCNGTNATGRGWGTNRTYGG-3′ and Cro2R: 5′-ANGAYTGRTARAANGANGAYTTYTT-3′) and L (outer: Han-L1880F: 5′-ATGAARNTNTGTGCNATNTTTGA-3′ and Han-L3000R: 5′-GCNGARTTRTCNCCNGGNGACCA-3′; inner: Han-L2520F: 5′-ATNWGHYTDAARGGNATGTCNGG-3′ and Han-L2970R: 5′-CCNGGNGACCAYTTNGTDGCATC-3′). Oligonucleotide primers designed for amplification and sequencing of the full genome of ARTV are shown in Table 2. Nested PCR cycling conditions and methods for DNA sequencing have been previously described (Arai et al. 2008a, 2008b, Kang et al. 2009b).
Mixed bases: B = G, T, C; D = G, A, T; H = A, T, C; K = G, T; M = A, C; N = A, T, G, C; R = A, G; W = A, T; and Y = C, T.
Genetic and phylogenetic analysis
Pairwise alignment and comparison of partial- and full-length S, M, and L segment sequences of hantaviruses from Laxmann's shrews, as well as other representative rodent-, shrew-, mole-, and bat-borne hantaviruses, were performed using the ClustalW method (Thompson et al. 1994). Phylogenetic analyses were conducted using maximum likelihood and Bayesian methods, implemented in MrBayes 3.1 (Ronquist and Huelsenbeck 2003) and RAxML (Stamatakis et al. 2008), under the best-fit GTR + I + Γ model of evolution using MrModeltest 2.3 (Posada 2008). Two replicate Bayesian Metropolis–Hastings Markov chain Monte Carlo runs, each consisting of six chains of 10 million generations sampled every 100 generations with a burn-in of 25,000 (25%), resulted in 150,000 trees overall. Topologies were evaluated by bootstrap analysis of 100 iterations, and posterior node probabilities were based on 10 million generations and estimated sample sizes more than 100 (implemented in MrBayes).
Host species identification
Because shrews are inherently difficult to identify by morphological features alone, host verification of ARTV-infected shrews was confirmed by analyzing the entire 1140-base pair cytochrome b gene of mitochondrial DNA (mtDNA), amplified by PCR, using modified universal primers (Cy-14726F: 5′-GACYARTRRCATGAAAAAYCAYCGTTGT-3′ and Cy-15909R: 5′-CYYCWTYIYTGGTTTACAAGACYAG-3′) (Arai et al. 2008b). A Bayesian approach, as described above, with midpoint rooting was used to determine the phylogenetic relationship between Laxmann's shrews and other shrew and mole species known to harbor distinct hantaviruses.
Results and Discussion
ARTV RNA was detected by RT-PCR in five of 39 (12.8%) and one of 29 (3.4%) Laxmann's shrews captured in Russia and Japan, respectively (Table 1). All but one of the six ARTV-infected shrews were males.
Analysis of the full-length nucleotide and amino acid sequences of the S, M, and L segments (GenBank accession numbers: KF974360, KF974359, and KF974361, respectively) of ARTV strain Mukawa AH301 showed considerable divergence from representative hantaviruses harbored by shrews in the Soricinae subfamily in Eurasia (Table 3): S, 22.9–27.3% and 13.3–16.0%; M, 20.9–23.8% and 8.4–12.7%; and L, 20.2–22.1% and 5.7–8.7%, respectively. In contrast, alignment and comparison between ARTV strain Mukawa AH301 and the very limited S, M, and L segments of ARTV strains ART502, Galkino2712, and Parnaya1205 showed higher sequence similarity at the amino acid level (Table 3).
For virus names and hosts, refer to legend of Figure 2.
Complete coding region.
—, sequences unavailable; aa, amino acids; ARTV, Artybash virus; nt, nucleotides; SWSV, Seewis virus.
Phylogenetic analyses of ARTV strain Mukawa AH301, based on the full-length coding regions (comprising 1290-nucleotide S, 3420-nucleotide M, and 6456-nucleotide L segment sequences), showed similar topologies, with ARTV strains from the Sakha Republic and ARTV strains detected independently in Laxmann's shrews captured in the Altai Republic and Khabarovsk Krai of Russia forming a separate geographic-specific cluster (Fig. 2). Moreover, ARTV strains were distinct from recently detected hantaviruses harbored by other Sorex shrew species in Eurasia. In contrast, differences in the phylogenetic positions of some soricine shrew-borne hantaviruses may be attributed to the limited sequences, especially for the M and L segments.
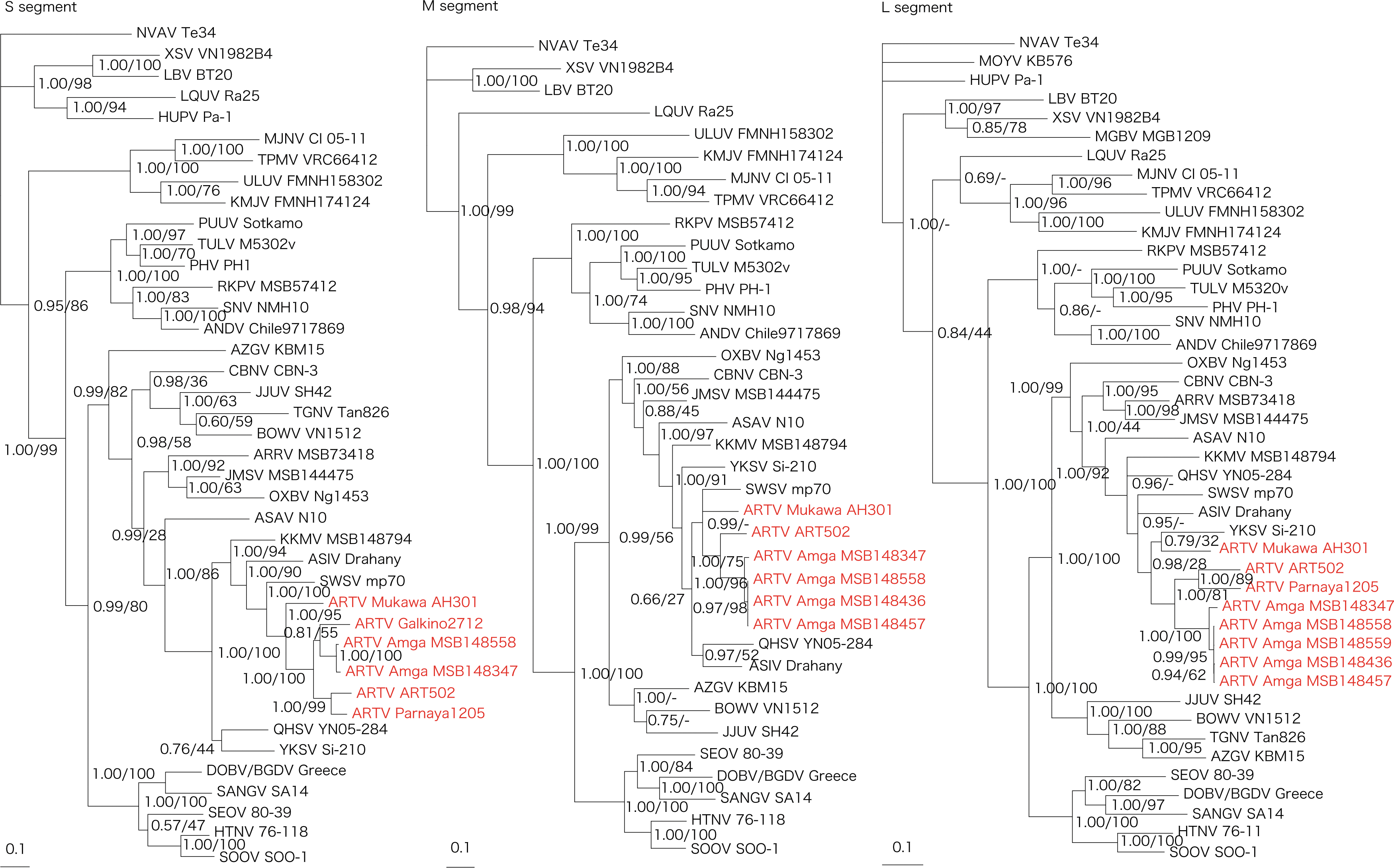
Phylogenetic trees generated by Bayesian and maximum likelihood methods under the best-fit GTR + I + Γ model of evolution as estimated based on the entire coding regions of the full-length 1290-nucleotide S, 3420-nucleotide M, and 6456-nucleotide L genomic segments of Artybash virus (ARTV) strain Mukawa AH301. Phylogenetic trees show the positions of ARTV Mukawa AH301 (GenBank accession numbers: S: KF974360; M: KF974359; and L: KF974361), ARTV Amga MSB148558 (S: KM201411; M: KM201412; and L: KM201413), ARTV Amga MSB148559 (S: KM201414), ARTV Amga MSB148436 (M: KM201415 and L: KM201416), ARTV Amga MSB148457 (M: KM201417 and L: KM201418), ARTV Amga MSB148347 (S: KM201419; M: KM201420; and L: KM201421), ARTV ART502 (S: KM288698; M: EU424340; and L: EU424339), ARTV Galkino2712 (S: KM288699), and ARTV Parnaya1205 (S: KU253274 and L: KU253275) from Sorex caecutiens. Also shown are Ash River virus (ARRV MSB73418, S: EF650086 and L: EF619961) from Sorex cinereus, Jemez Springs virus (JMSV MSB144475, S: FJ593499; M: FJ593500; and L: FJ593501) from Sorex monticolus, Seewis virus (SWSV mp70, S: EF636024; M: EF636025; and L: EF636026) from Sorex araneus, Asikkala virus (ASIV Drahany, S: KC880342; M: KC880345; and L: KC880348) from Sorex minutus, Kenkeme virus (KKMV MSB148794, S: GQ306148; M: GQ306149; and L: GQ306150) from Sorex roboratus, Qian Hu Shan virus (QHSV YN05-284, S: GU566023; M: GU566022; and L: GU566021) from Sorex cylindricauda, Yakeshi virus (YKSV Si-210, S: JX465423; M: JX465403; and L: JX465389) from Sorex isodon, and Cao Bang virus (CBNV CBN-3, S: EF543524; M: EF543526; and L: EF543525) from Anourosorex squamipes, as well as Thottapalayam virus (TPMV VRC66412, S: AY526097 and L: EU001330) from Suncus murinus, Imjin virus (MJNV Cl 05-11, S: EF641804; M: EF641798; and L: EF641806) from Crocidura lasiura, Azagny virus (AZGV KBM15, S: JF276226; M: JF276227; and L: JF276228) from Crocidura obscurior, Tanganya virus (TGNV Tan826, S: EF050455 and L: EF050454) from Crocidura theresea, Bowé virus (BOWV VN1512, S: KC631782; M: KC631783; and L: KC631784) from Crocidura douceti, Jeju virus (JJUV SH42, S: HQ663933; M: HQ663934; and L: HQ663935) from Crocidura shantungensis, Uluguru virus (ULUV FMNH158302, S: JX193695; M: JX193696; and L: JX193697) from Myosorex geata, and Kilimanjaro virus (KMJV FMNH174124, S: JX193698; M: JX193699; and L: JX193700) from Myosorex zinki. Mole-borne hantaviruses include Asama virus (ASAV N10, S: EU929072; M: EU929075; and L: EU929078) from Urotrichus talpoides, Nova virus (NVAV Te34, S: KR072621; M: KR072622; and L: KR072623) from Talpa europaea, Oxbow virus (OXBV Ng1453, S: FJ5339166; M: FJ539167; and L: FJ593497) from Neurotrichus gibbsii, and Rockport virus (RKPV MSB57412, S: HM015223; M: HM015222; and L: HM015221) from Scalopus aquaticus. Bat-borne hantaviruses include Magboi virus (MGBV MGB1209, L: JN037851) from Nycteris hispida, Mouyassué virus (MOYV KB576, L: JQ287716) from Neoromicia nanus, Huangpi virus (HUPV Pa-1, S: JX473273 and L: JX465369) from Pipistrellus abramus, Longquan virus (LQUV Ra-25, S: JX465415; M: JX465397; and L: JX465381) from Rhinolophus sinicus, Laibin virus (LBV BT20, S: KM102247; M: KM102248; and L: KM102249) from Taphozous melanopogon, and Xuan Son virus (XSV VN1982B4, S: KC688335; M: KU976427; and L: JX912953) from Hipposideros pomona. Other taxa include Sin Nombre virus (SNV NMH10, S: NC_005216; M: NC_005215; and L: NC_005217), Andes virus (ANDV Chile9717869, S: AF291702; M: AF291703; and L: AF291704), Prospect Hill virus (PHV PH-1, S: Z49098; M: X55129; and L: EF646763), Tula virus (TULV M5302v, S: NC_005227; M: NC_005228; and L: NC_005226), Puumala virus (PUUV Sotkamo, S: NC_005224; M: NC_005223; and L: NC_005225), Dobrava virus/Belgrade virus (DOBV/BGDV Greece, S: NC_005233; M: NC_005234; and L: NC_005235), Hantaan virus (HTNV 76-118, S: NC_005218; M: NC_005219; and L: NC_005222), Soochong virus (SOOV SOO-1, S: AY675349; M: AY675353; and L: DQ056292), Sangassou virus (SANGV SA14, S: JQ082300; M: JQ082301; and L: JQ082302) and Seoul virus (SEOV 80-39, S: NC_005236; M: NC_005237; and L: NC_005238). The numbers at each node are posterior node probabilities (left) based on 150,000 trees: two replicate Markov chain Monte Carlo runs, consisting of six chains of 10 million generations each sampled every 100 generations with a burn-in of 25,000 (25%) and bootstrap values (right), based on 100 replicates executed on the RAxML BlackBox Web server.
The identities of the six ARTV-infected shrews were confirmed as Laxmann's shrews (GenBank no. KF974362 for strain AH301; GU562415, GU562416, GU562417, GU562418, and GU562422 for strains MSB148558, MSB148559, MSB148436, MSB148457, and MSB148347, respectively). The cytochrome b mtDNA sequences of the ARTV-infected Laxmann's shrews from Japan and Russia differed by 4.0–6.4%. The ARTV-negative shrews from Japan were also identified as Laxmann's shrews by analysis of the 1140-nucleotide cytochrome b mtDNA gene (GenBank nos. KU760858 to KU760884). However, the full-length cytochrome b mtDNA sequence variation was 0–1.1% and 0.5–1.1% among Laxmann's shrews in Japan and Russia, respectively.
Host phylogenies based on mtDNA cytochrome b sequences of shrew and mole species, which harbor genetically distinct hantaviruses, showed two separate lineages of Laxmann's shrews (Fig. 3). This was consistent with previous studies suggesting at least two genetic races of Laxmann's shrews distributed in Eurasia and Japan (Ohdachi et al. 2003). That is, based on analysis of the full-length cytochrome b gene, Laxmann's shrews were separated into Hokkaido and Eurasian Continent–Sakhalin–Cheju clusters. Individuals in the latter cluster do not always reflect the geographical proximity of their capture locations, which is consistent with an ancestral isolation of the Hokkaido population, occurring ∼13,000 years before present (Tanabe et al. 2015), and recent rapid range expansion of the modern Eurasian Continent–Sakhalin–Cheju population (Ohdachi et al. 2003).
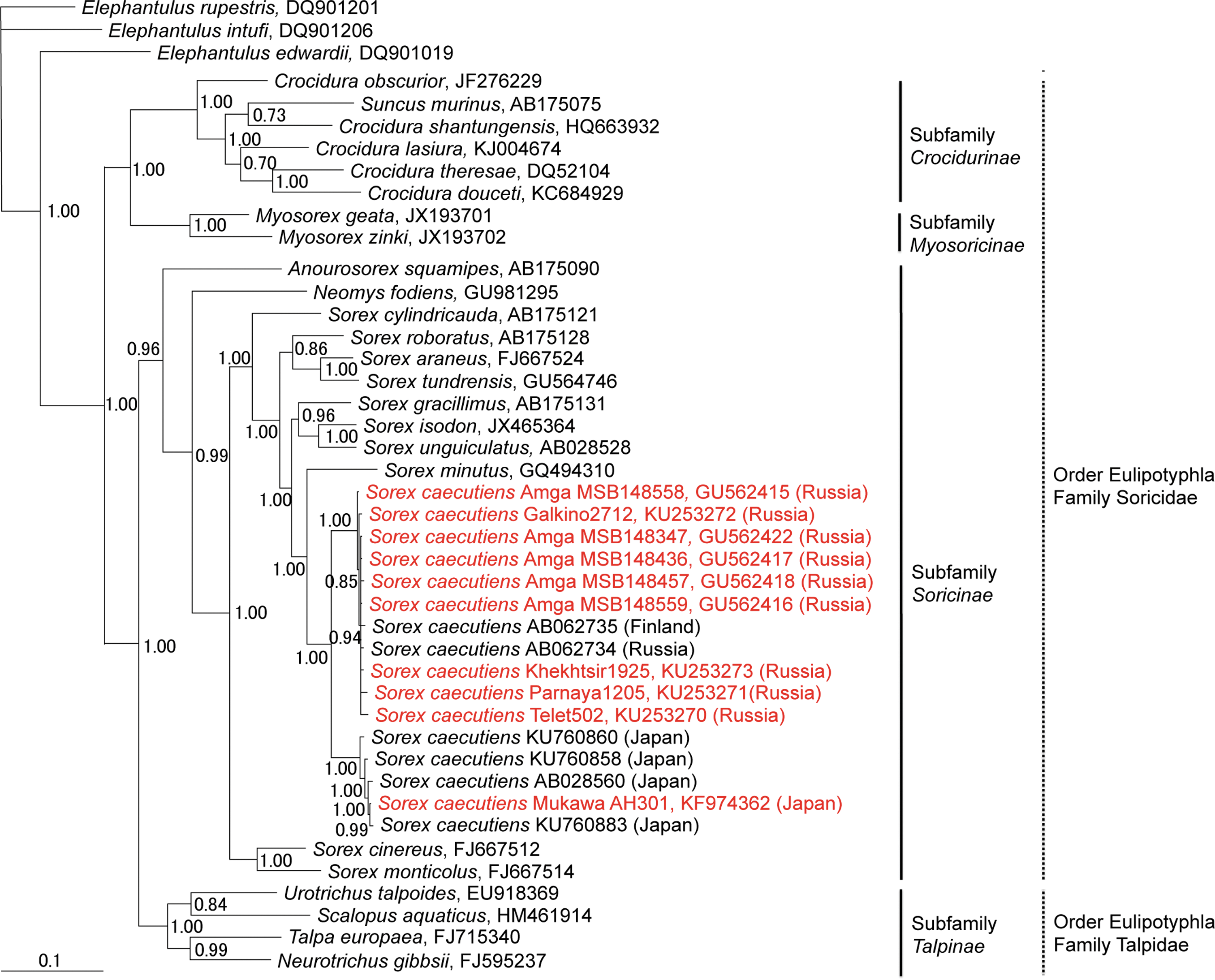
Bayesian phylogenetic tree based on 1140 nucleotides of the cytochrome b mtDNA of shrews and moles (order Eulipotyphla and families Talpidae and Soricidae). The tree was rooted using Elephantulus (order Macroscelidea, GenBank nos. DQ901019, DQ901206, and DQ901201) as the outgroup. Numbers at nodes indicate posterior probability values based on 150,000 trees: two replicate Markov chain Monte Carlo runs, consisting of six chains of 10 million generations each sampled every 100 generations with a burn-in of 25,000 (25%). GenBank numbers for all taxa are provided in the tree.
The phylogeographic variation in ARTV reflects the distinct evolutionary histories of these variants. Similar patterns of geographic variation have also been reported for rodent-borne hantaviruses. For example, geographic-specific genetic variants have been reported for Puumala virus in the bank vole (Myodes glareolus) (Garanina et al. 2009), Tula virus in the European common vole (Microtus arvalis) (Song et al. 2004), and Andes virus in the colilargo (Oligoryzomys longicaudus) (Torres-Perez et al. 2011). Both Muju virus in the royal vole (Myodes regulus) (Lee et al. 2014) and Hokkaido virus in the grey red-backed vole (Myodes rufocanus) and northern red-backed vole (Myodes rutilus) (Yashina et al. 2015) may represent genotypes of Puumala virus. These examples point to the urgency of studying the phylogeographic variation of viruses and their hosts to provide an essential foundation for understanding their evolutionary histories and as a prelude to forecasting their future emergence under changing environmental conditions (Hope et al. 2013, Campbell et al. 2015).
Although ARTV was detected in a single Laxmann's shrew captured in the Mukawa area in Japan, the evidence is compelling that this Sorex shrew species harbors ARTV across its broad geographic range. Our findings suggest that Laxmann's shrews, resident on Hokkaido Island before the geologic separation from the Eurasian continent, might have already been infected with ARTV. Renewed attempts to isolate ARTV from Laxmann's shrews captured in Eurasian Continent–Sakhalin–Cheju and Japan are warranted to better understand its evolutionary origins and phylogeography, as well as its pathogenic potential in humans.
Footnotes
Acknowledgments
We thank Masashi Hamada, Yu Ikeyama, and Keita Aoki for their technical assistance and Kimiyuki Tsuchiya, Hidenori Nishizawa, and Kyle R. Taylor for supporting field investigations. We also thank Nikolai Dokuchaev, Andrew G. Hope, Anson Koehler, Stephen O. MacDonald, Robert A. Nofchissey, and Albina Tsvetkova for assistance with field collections made in Russia through the National Science Foundation support (DEB0415668). In addition, this research was supported in part by a grant-in-aid for Research on Emerging and Re-emerging Infectious Diseases, Health Labour Sciences Research Grant in Japan (H22-Shinko-Ippan-006 and H25-Shinko-Ippan-008), a grant-in aid for Research Program on Emerging and Re-emerging Infectious Diseases, Japan Agency for Medical Research and Development (AMED), and a grant-in-aid from the Japan Society for the Promotion of Science (S13205 Invitation Fellowship for Research in Japan), as well as by the U.S. Public Health Service grants R01AI075057 from the National Institute of Allergy and Infectious Diseases and P20GM103516 and P30GM114737 (Centers of Biomedical Research Excellence) from the National Institute of General Medical Sciences, National Institutes of Health.
Author Disclosure Statement
No competing financial interests exist.