
Abstract
Helicobacter species are newly emerging bacteria with great public implications but till now its epidemiology is not fully understood; so, this study was conducted to investigate the possible role of ruminants in the epidemiology of these pathogens. For this purpose, fecal samples were collected from 149 animals (76 sheep, 33 goats, 21 cattle, and 19 buffaloes) and stool specimens from 10 animal caretakers in intimate contact with the examined animals. All samples were examined for the presence of Helicobacter species through detection of Helicobacter genus specific 16S rRNA using PCR. Then, all positive Helicobacter spp. amplicons were sequenced to recognize their species through BLAST analysis at GenBank. The overall prevalence of Helicobacter spp. was 14.8% while the distribution among the different animals was 26.3%, 3%, 4.8%, and 0% in sheep, goats, cattle, and buffaloes respectively. Helicobacter canis was the predominant species and detected only in sheep (21%) and goats (3%). Moreover, Helicobacter winghamensis and Helicobacter canadensis were also detected in sheep but not in other animals, whereas the only positive bovine sample was identified as Helicobacter bovis. On the other hand, 4 out of 10 humans were positive for Helicobacter spp. and all sequences were identified as H. canis. The sequences identity matrix and phylogenetic analysis of H. canis sequences from humans and sheep contacts revealed that one human sequence was identical to that of sheep and making sister group clade, which prove the zoonotic transmission of this pathogen between sheep and human contacts. However, our findings highlight sheep as a potential reservoir for H. canis, further researches are needed to address the potential role of sheep in the food-borne transmission of such emerging pathogen.
Introduction
T
Materials and Methods
Fecal samples were collected from 76 sheep, 33 goats, 21 cattle, and 19 buffaloes (apparently healthy, randomly selected from different farms in Giza and Sharkia governorates, Egypt), all samples were obtained from rectum of the examined animals using sterile gloves and placed in sterile cups. Moreover, stool specimens were gathered from 10 animal caretakers (apparently healthy) in intimate contact with the examined animals through their voluntary contribution in the study. All samples were transported in icebox to laboratory of Department of Zoonoses, Faculty of Veterinary Medicine, Cairo University for further processing.
Molecular detection of Helicobacter spp
All examined animals and humans were tested for the presence of Helicobacter spp. in their feces through direct detection of Helicobacter genus specific16S rRNA gene by PCR.
Extraction of DNA
DNA was extracted from fecal and stool samples of examined animals and humans using QIAamp DNA stool mini kit (Qiagen) according to the procedure of the kit. The extracted DNA was stored at −20°C until use.
Polymerase chain reaction
PCR was done upon the extracted DNAs to detect 16S rRNA gene specific for genus Helicobacter using the following pair of primers:
Forward: 5′ GGCTATGACGGGTATCCGGC 3′ and reverse 5′ GCCGTGCAGCACCTGTTTTC 3′. The primers were synthesized by (Metabion) according to (Moyaert et al. 2008). The amplification reaction was carried out using HotStarTaq Master Mix kit (Qiagen). The thermal profile of the reaction was initial denaturation at 95°C for 15 min, 45 cycles of denaturation at 95°C for 30 s, annealing at 65°C for 30 s; extension at 72°C for 30 s. Then final extension for 10 min at 72°C. Afterward, the amplicons were entered electrophoresis step and specific bands were noted at 764 bp (Fig. 1) (Moyaert et al. 2008).
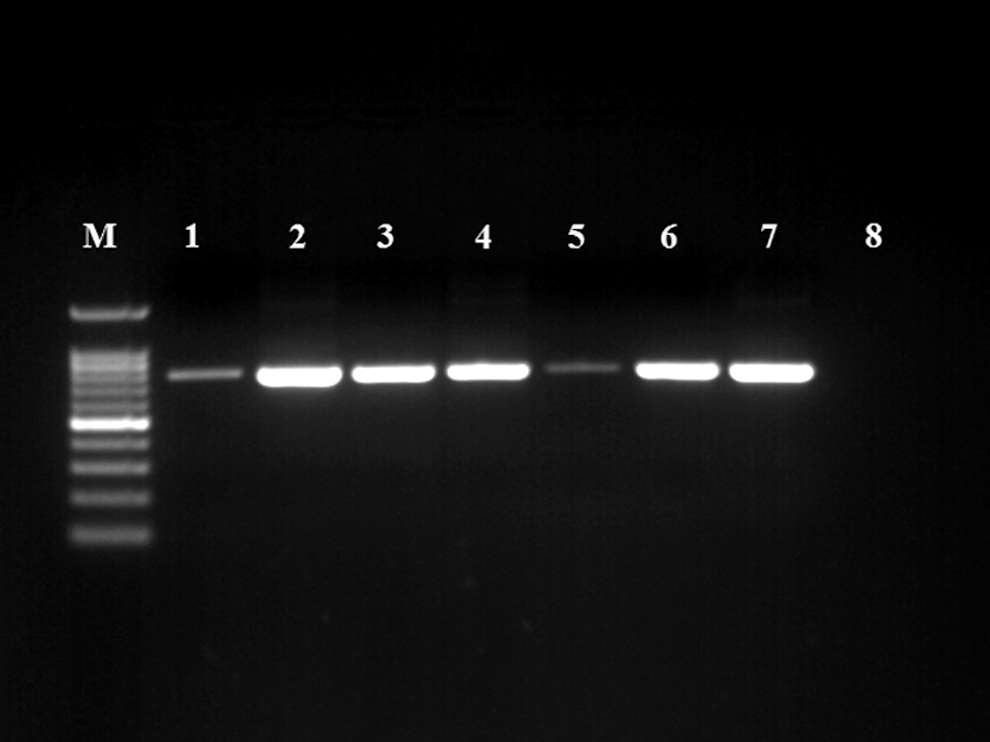
Occurrence of Helicobacter spp. among examined animals and humans Lane M: DNA ladder; lanes 1–7: positive samples for Helicobacter spp. showed specific bands at 764 bp; lane 8: negative control.
Gene sequencing for 16S rRNA gene
PCR products of positive samples were purified using GeneJET™ PCR purification kit (Thermo) and the sequencing step was conducted with Big Dye Terminator V3.1 Cycle sequencing Kit (Applied Biosystems). The obtained sequences were identified through BLAST analysis with sequences available at GenBank.
Sequences identity matrix and phylogenetic analysis of Helicobacter canis from humans and sheep contacts
All human sequences were aligned against Helicobacter canis sequences from contact sheep and against the most similar sequences available at GenBank to study the identity and evolutionary history of these sequences. Clustral W Multiple alignment and sequences identity matrix were done with using (BioEdit) software whereas phylogenetic tree was constructed by neighbor-joining method using MEGA6 software version (6.06) (Fig. 2).

Phylogenetic tree constructed by neighbor-joining method with boot-strap test (replicates 500). The analysis was conducted based on partial sequence of 16S rRNA gene of 20 Helicobacter canis sequences. The analysis was done by MEGA6 version (6.06).
Nucleotide sequence accession numbers
The nucleotide sequences of 16S rRNA of H. canis determined in this study have been deposited in GenBank under the following accession numbers:
H. canis from humans: KX028790-KX028792; KX036662.
H. canis from sheep: KX037371; KX064408-KX064417.
Results
Of 149 examined animals, 22 yielded Helicobacter spp. with an overall prevalence 14.8%. The occurrence of Helicobacter spp. among different animal species was 26.3%, 3%, 4.8%, and 0% in sheep, goats, cattle, and buffaloes, respectively (Table 1). The results of 16S rRNA sequencing of the amplicons identified most of detected Helicobacter spp. to be H. canis with the following prevalence among different animal species 21%, 3% for sheep and goats respectively while none of cattle yielded H. canis. Moreover, Helicobacter canadensis and Helicobacter winghamensis were also identified in sheep with a prevalence of 2.6% for each, whereas the bovine sequence was identified as Helicobacter bovis (Table 1). On the other hand, all human Helicobacter sequences were identified as H. canis (Table 1). The sequences identity matrix of H. canis from both human and sheep contacts and 16S rRNA sequence of H. canis strain ATCC 51401 were displayed in (Table 2).
Discussion
In the recent years genus Helicobacter has gained ground in the medical community as it stands behind many gastrointestinal complaints in humans. The results of this study revealed the occurrence of Helicobacter spp. in the feces of the examined animals in an overall prevalence 14.8% whereas the prevalence rates among different animal species showed that the vast majority of positive results were derived from sheep (26.3%) rather than the other examined animals. Furthermore, the sequencing results of 16S rRNA revealed the predominance of H. canis (21%) rather than other Helicobacter spp. (H. canadensis, H. winghamensis) (2.6% for each) among examined sheep. However, H. canis was first recognized in dogs and cats (Stanley et al. 1993, Foley et al. 1999), a recent study by Swennes et al. (2014) isolated H. canis from sheep to shed light on sheep as a possible reservoir for H. canis. The results of our study augment this concept and underscore sheep as a potential reservoir for H. canis and may play a pivotal role in its epidemiology through shedding of the pathogen in its feces to contaminate milk, animal carcass, and may be transmitted directly through fecal-oral route to human contacts. Interestingly, the results of human samples provide concrete evidence on the zoonotic transmission of H. canis between sheep and human contacts as out of 10 animal caretakers, 4 were positive for Helicobacter spp. while all human sequences were identified as H. canis. All H. canis-positive humans were in intimate contact with 11 positive sheep and lived in the same farm. On the molecular basis, one human sequence was identical to one ovine sequence and making a sister group as revealed by phylogentic tree and 100% identity as appeared in sequence identity matrix, whereas other human sequences showed high genetic relatedness with the animal ones. To the best of our knowledge, this is the first record of zoonotic transmission of H. canis between sheep and human contacts, which highlights the possible role of sheep in the epidemiology of H. canis. Moreover, the sequences identity matrix and the phylogentic analysis of the obtained H. canis from sheep and human contacts with other published H. canis sequences showed high level of identity and some sequences were grouped in the same clade with those previously recorded in serious human cases such as bacteremia in patient with fever of unknown origin (Abidi et al. 2013) and bacteremia in patient with multifocal cellulitis a matter that magnifies the public health burden of our findings. Additionally, we could identify both emerging enteropathogenic Helicobacter spp.; H. canadensis and H. winghamensis from sheep feces, which are responsible for gastroenteritis in humans (Melito et al. 2001, Waldenstrom et al. 2003, Loman et al. 2009), raising the probability of sheep to be a zoonotic reservoir for such pathogens. Despite the large number of helicobacters that could be identified in sheep feces, we only detected Helicobacter sp. in one goat and identified as H. canis. Nonetheless, to the best of our knowledge it is the first time H. canis was detected in the feces of goats. On the other hand, Helicobacter sp. was detected only once in cattle and recognized as H. bovis while none of the examined buffaloes yielded positive results. In conclusion, the detection of H. canis in sheep feces in such high prevalence expands this pathogen host and identifies sheep as a potential reservoir for H. canis. Moreover, this study records a zoonotic transmission between sheep and human contacts, which is new evidence in the epidemiology of such pathogen.
Footnotes
Author Disclosure Statement
No competing financial interests exist.