
Abstract
Usutu virus (USUV) is a mosquito-borne flavivirus accounting for large-scale deaths in resident bird populations. In this study, we show the introduction of USUV to Eastern Germany resulting in massive death of birds, particularly blackbirds (Turdus merula). We found that three diverse USUV lineages (“Europe 3,” “Africa 2,” and “Africa 3-like”) circulated simultaneously. Moreover, we detected USUV in Culex pipiens in a region where no dead birds were reported, strengthening the need for mosquito monitoring to uncover the spread of arboviruses. Furthermore, phylogenetic analyses revealed that mutations accumulated, in particular, in the NS3 region within short time periods. In addition, comparison of whole-genome sequences showed that diverse isolates of the cluster “Africa 3-like” are cocirculating in Germany due to independent introduction events.
Introduction
U
Results and Discussion
Between August and September 2016, numerous dead blackbirds were found in a 100-km zone around the city of Leipzig, Eastern Germany (Fig. 1A). Histopathological examinations showed that most of them suffered from necrotizing splenitis, hepatitis, and acute encephalitis, hallmarks of both USUV and WNV infection. Therefore, we determined the presence of USUV and WNV as described previously (Linke et al. 2007, Jöst et al. 2011) in RNA isolated from the liver of dissected birds (RNeasy Mini Kit; Qiagen). All samples were negative for WNV, but 15 out of 23 birds were USUV positive. All of them were blackbirds (Turdus merula), except for one great gray owl (Strix nebulosa) found in the zoo of the city of Halle (Saale) (Fig. 1A).
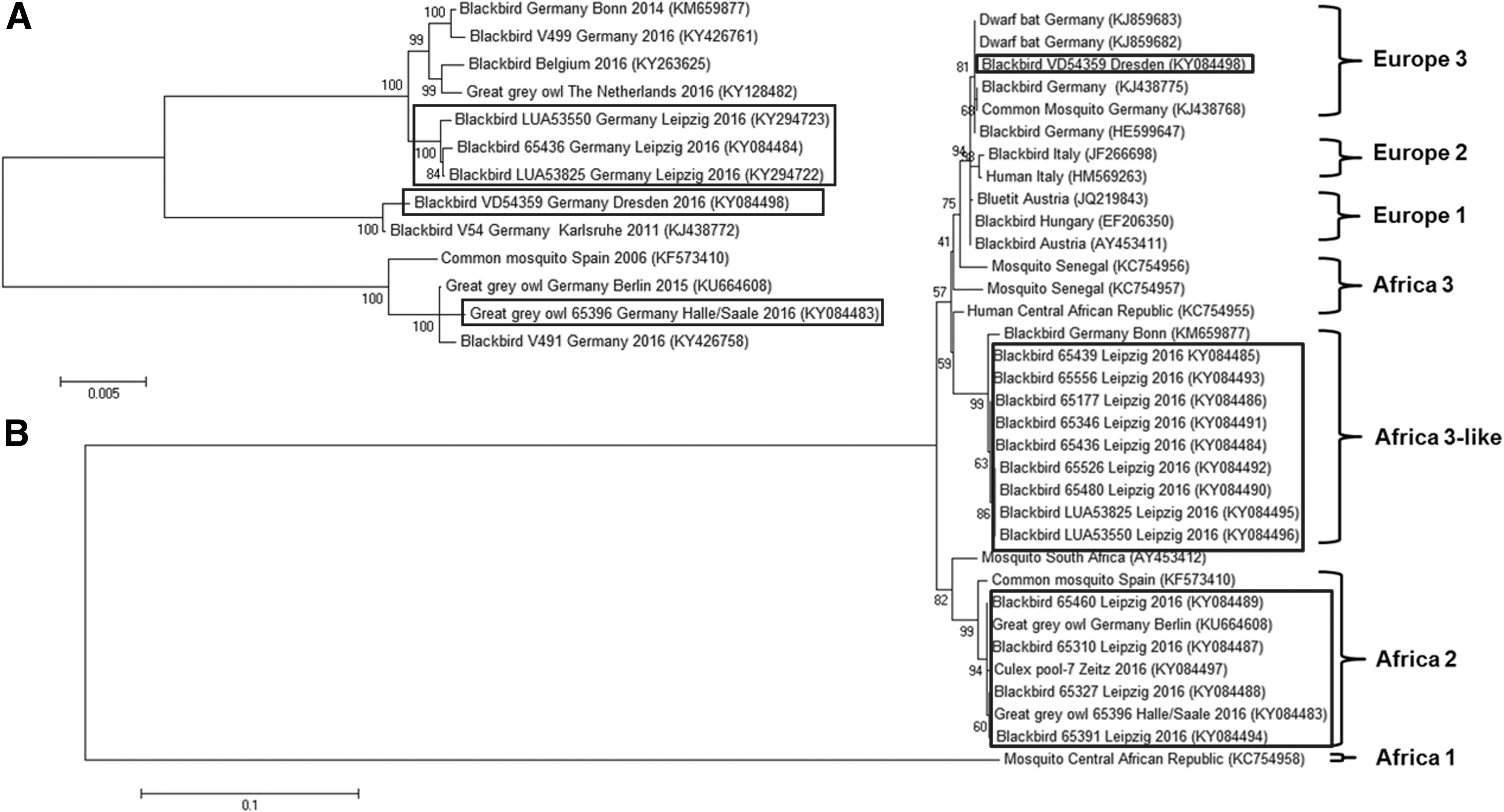
Phylogeny of detected USUV isolates.
To find out when USUV was first introduced to the affected region, we retrospectively analyzed 25 birds from this area, collected between 2009 and 2015, for USUV and WNV. All samples were negative for both viruses, suggesting that the introduction event took place in 2016, although this could not be proved finally due to limited available numbers of bird samples. To clarify the distribution of USUV, we also trapped mosquitoes around Leipzig (animals were trapped by hand over a period of 6 weeks using sterile plastic containers followed by immediate freezing at −20°C) and analyzed pools of 10 animals belonging to the Culex pipiens complex for USUV and WNV RNA as described above. A total number of 13 pools were investigated, whereby one pool was found to be positive for USUV, but all were WNV negative. The USUV-positive mosquitoes were collected from a household in the city of Zeitz, which is in the distance of 50 km from Leipzig. No dead birds were reported in that region (Fig. 1A). This finding reflects that the prevalence of USUV should be determined by using mosquitoes as well as wild and captive birds as indicator animals.
To uncover how USUV was introduced into Eastern Germany, we performed phylogenetic analysis of all isolated strains using PCR protocols established by Cadar et al. (2015). Amplifications were set up using the “SuperScript III One-Step RT-PCR System with Platinum Taq High Fidelity” (Life Technologies). PCR fragments were purified with a PCR DNA Fragments Extraction Kit (Geneaid) followed by Sanger sequencing (Seqlab). Nucleotide sequences were then analyzed using the Basic Local Alignment Search Tool (BLAST,
Nucleotide sequence alignment of a 915 bp fragment coding for parts of the envelope protein revealed that the isolated strains matched to the USUV clusters “Europe 3,” “Africa 2,” and “Africa 3-like” (Fig. 1B). Highest similarity was found in strains previously isolated in other parts of Germany. Based on the analyzed envelope protein fragment, nucleotide similarity between isolated strains grouping to the same USUV cluster was nearly 100% identical. For this reason, an in depth analysis of representative isolates of each cluster was performed by whole-genome sequencing (Ziegler et al. 2016) and alignment with the above-mentioned German and European isolates of highest similarity. This analysis revealed that especially the genome region coding for the NS3 protein constituted a hotspot for mutations in all three clusters. This makes the NS3 region particularly suitable for epidemiological studies of regional outbreaks.
In addition, comparison of whole-genomic sequences revealed that the Leipzig isolates belonging to the “Africa 3-like” cluster can be clearly distinguished from previously detected strains from Germany (Bonn, 2014; accession no. KM659877 and V499, 2016, accession no. KY426761), Belgium (2016, accession no. KY263625), and the Netherlands (2016, accession no. KY128482). Therefore, an independent introduction event of these local USUV-isolates can be assumed, highlighting the cocirculation of diverse “Africa 3-like” strains in different parts of Germany in 2016 (Fig. 1A). Cadar et al. (2015) proposed a new lineage based on the Bonn isolate, which is now further supported by our data since these strains can be easily separated from the classical “Africa 3” ones (Fig. 1B).
In contrast, our “Europe 3” and “Africa 2” isolates exhibited fewer nucleotide changes when compared with their respective closest relatives from Germany (Fig. 1A). This observation may be explained due to the establishment of these USUV-lineages in the affected regions from former introductions. Further investigations are needed to stress this hypothesis and to uncover the detailed epidemiological situation in Germany.
The cocirculation of different USUV lineages in one region raises the concern that recombination events could occur and speed up the diversity of USUV. Recombination between genomes of the Japan encephalitis virus complex, which also comprises USUV, were reported, at least in laboratory conditions (Taucher et al. 2010). This finding and the results of our study stress that further surveillance is urgently needed to characterize the fast spread of USUV, in particular, the novel variants of the “Africa 3-like” lineage.
Footnotes
Acknowledgments
We thank Katrin Erfurt and Jana Schömburg for perfect technical assistance and Bastian Thaa for critical reading of the article. Nucleotide sequences accession numbers: KY084483, KY084484, KY084485, KY084486, KY084487, KY084488, KY084489, KY084490, KY084491, KY084492, KY084493, KY084494, KY084495, KY084496, KY084497, KY084498, KY199556, KY199557, and KY199558.
Author Disclosure Statement
No competing financial interests exist.