
Abstract
Japanese encephalitis virus (JEV) is a representative virus of the JEV serogroup in genus Flavivirus, family Flaviviridae. JEV is a mosquito-borne virus that causes Japanese encephalitis (JE), one of the most severe viral encephalitis diseases in the world. JEV is divided into five genotypes (G1–G5), and each genotype has its own distribution pattern. However, the distribution of different JEV genotypes has changed markedly in recent years. JEV G1 has replaced G3 as the dominant genotype in the traditional epidemic areas in Asia, while G3 has spread from Asia to Europe and Africa and caused domestic JE cases in Africa. G2 and G5, which were endemic in Malaysia, exhibited great geographical changes as well. G2 migrated southward and led to prevalence of JE in Australia, while G5 emerged in China and South Korea after decades of silence. Along with these changes, JE occurred in some non-traditional epidemic regions as an emerging infectious disease. The regional changes in JEV pose a great threat to human health, leading to huge disease burdens. Therefore, it is of great importance to strengthen the monitoring of JEV as well as virus genotypes, especially in non-traditional epidemic areas.
Introduction
J
JEV is a representative virus of the JEV serogroup in genus Flavivirus, family Flaviviridae. JEV is a single-stranded, positive-sense RNA virus. Its genome is about 11 kb in length, containing only one open reading frame (ORF) that encodes a polyprotein. The genome encodes three structural proteins, namely capsid (C), membrane (M), and envelope (E) proteins, as well as seven non-structural proteins (NS1, NS2a, NS2b, NS3, NS4a, NS4b, and NS5). There are 5′ and 3′ non-coding regions (NCRs) at the ends of the ORF region. The structural proteins of JEV construct an icosahedral nucleocapsid and glycated E with M inlaid in lipid bilayers. E protein participates in many essential biological processes, including hemagglutination, neutralization of viruses, and assembly of viral particles. The viral non-structural proteins provide functional regulatory proteases for JEV duplication (Sumiyoshi et al. 1987, Lindenbach et al. 2007).
JEV is divided into five genotypes (G1–G5) according to molecular genetic analysis of their genomic sequences. The geographical distribution pattern of each genotype differs. For example, JEV G1 is mainly endemic in the tropics, while G3 is found in temperate regions (Uchil and Satchidanandam 2001, Solomon et al. 2003). However, the geographic distribution of each JEV genotype has changed markedly since the beginning of the 21st century. For instance, JEV G1 is not only endemic in Southeast Asia, but has also gradually spread northward to invade vast areas of South, Central, and East Asia, and has become the dominant genotype in these regions, displacing G3 (Nga et al. 2004, Wang et al. 2007a, Nitatpattana et al. 2008, Pan et al. 2011). A study published in 2014 revealed that JEV G1 was divided into two large clades, GI-a and GI-b. GI-a was the clade continuously prevalent in the tropical regions of Asia, while GI-b was the one now widespread and gradually replaced G3 as the dominant genotype in Asia (Schuh et al. 2014). In addition, G3 was isolated in specimens of mosquitoes and birds in southern Europe, a non-epidemic area of JEV (Platonov et al. 2012, Ravanini et al. 2012). Moreover, a local JE case was reported due to G3 JEV infection in Angola, Africa in 2016 (Simon-Loriere et al. 2017). These occurrences indicate that Europe and Africa may become new epidemic areas for JE. Therefore, the changing geographic distribution of JEV genotypes and the prevalence of related diseases are not only scientific questions of concern to virologists, but also public health issues related to human life. Thus, we discuss the characteristics of the changing geographic distribution of different JEV genotypes and the relationship with the prevalence of viral encephalitis, aiming to provide baseline data for global assessment of JEV risk and the prevalence of JE.
JEV Genotypes and Their Geographic Distributions
Analysis of the JEV genome showed that the nucleotide sequence between 456 and 695 bases (240 nt in length) in the PrM region was conserved. A nucleotide difference in this sequence usually occurred in the third position of the amino acid codon (silent mutation) and thus the amino acid sequence was stable. Therefore, this sequence is suitable for use in phylogenetic analysis due to evolutionary pressure on the region. Based on this theory, genotyping of JEV was first proposed in 1990. Subsequently, molecular genetic analysis was conducted using all JEV strains registered in GenBank, and the results indicated that the JEVs clustered into four independent branches, or four genotypes (G1–G4) (Chen et al. 1990). Later, JEV G5 was identified using the same method (Chen et al. 1992).
Although JEV was divided into five genotypes according to the conserved sequence of the PrM region, this sequence is too short (240 nt) to represent the entire genome, as it conveys much less information than the whole 11-kb genome. The E gene is the most important structural gene of JEV, and the nucleotide sequence of the E gene represents the interaction of the virus with host cells and the pathogenicity of JEV. Therefore, the sequence of the E gene has been suggested as a molecular basis for viral genotyping. Subsequent studies revealed that JEV genotyping results using the E gene were identical to those using the 240-nt sequence of the PrM gene (Ni and Barrett 1995, Mangada and Takegami 1999). With the development of sequencing techniques, it is becoming easier to obtain whole-genome sequences, making it possible to use the entire sequence as the basis of viral genotyping. Related research demonstrated that JEV could also be divided into five genotypes based on whole-genome sequences (Solomon et al. 2003, Pan et al. 2011).
A previous study revealed the differing patterns of geographic distribution of the JEV genotypes (Solomon et al. 2003). JEV G1–G5 coexisted in the tropical Indonesia-Malaysia region. Australia and New Guinea contained G1 and G2. The Philippines had G2 and G3. Thailand, Cambodia, and Vietnam had G1, G2, and G3. Japan, the Korean Peninsula region and most parts of China had G1 and G3, while South Asia including India, Sri Lanka, and Nepal had only G3 (Fig. 1). Because G1–G5 overlapped in the Malaysia-Indonesia tropical area, while all other countries and regions have only one or two genotypes, JEV is considered to have originated in the Malaysia-Indonesia region (Solomon et al. 2003). Subsequent studies have supported this conclusion (Gao et al. 2013, Schuh et al. 2014).
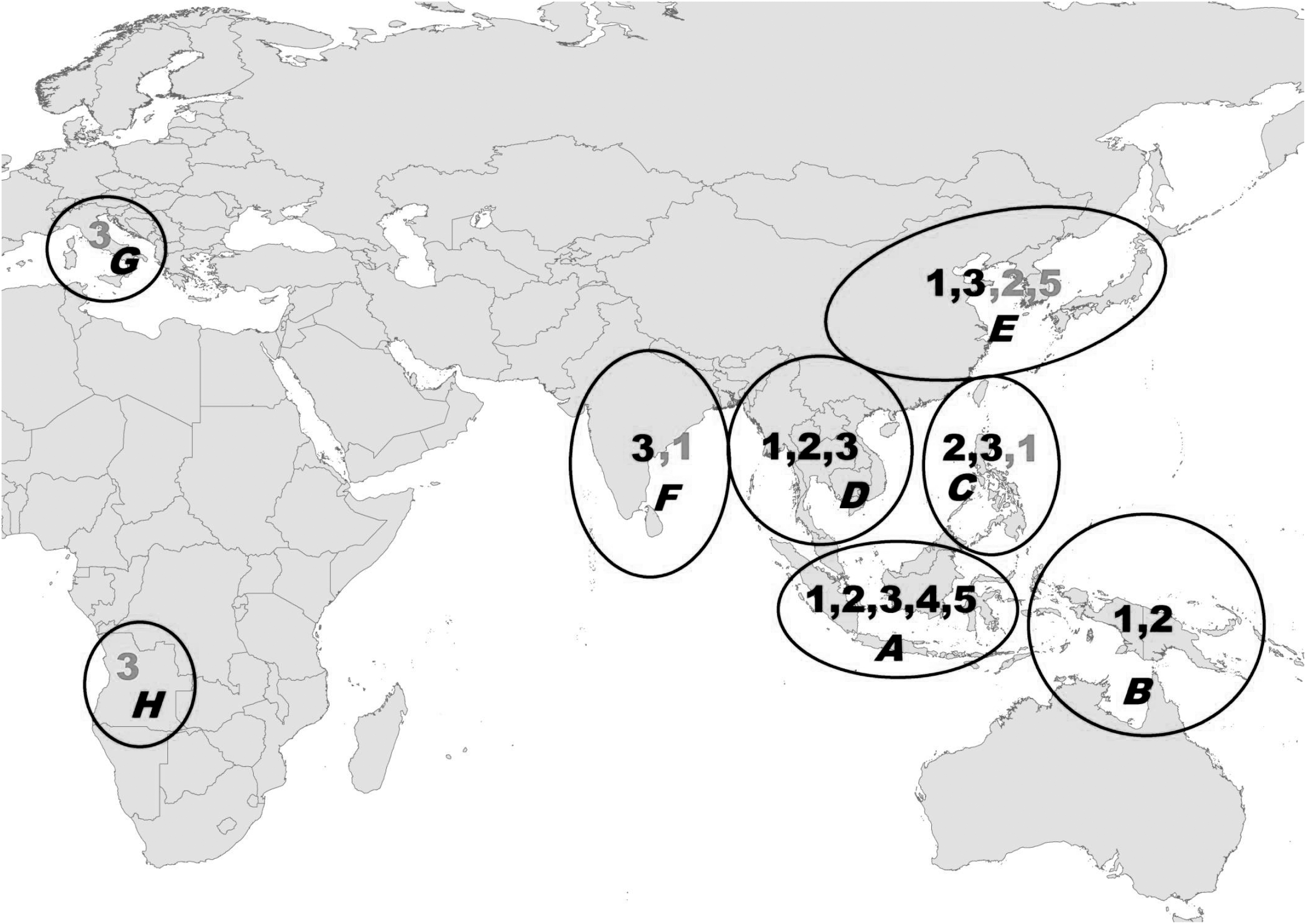
Geographic distribution of JEV genotypes 1–5. A–F: traditional epidemic areas of JEV and JE in Asia and Oceania. A, Indonesia (excluding New Guinea) and Malaysia; B, Australia and New Guinea; C, Taiwan (China) and the Philippines; D, Thailand, Cambodia, Vietnam, Laos, and Myanmar; E, China, Japan, and South Korea; and F, India, Sri Lanka, and Nepal. G and H: Emerging JE areas that were previously non-epidemic areas of JEV and JE. G: Italy; H, Angola. The numbers in each circle are JEV genotypes that have become endemic in the region. The numbers in black are JEV genotypes prevalent in these regions (A–F) before 2000, while gray numbers are JEV genotypes emerged after 2000. JEV, Japanese encephalitis virus.
In this study, we collected all JEV sequences (after removing duplicate sequences, derivative sequences, etc.) registered in GenBank as of December 19, 2017 for analysis and summarized the distribution of JEV genotypes according to time period, as well as by countries and regions (Table 1 and Fig. 1). Additionally, to demonstrate the geographical distribution of different genotypes, representative strains were selected and a phylogenetic tree was prepared (Fig. 2).
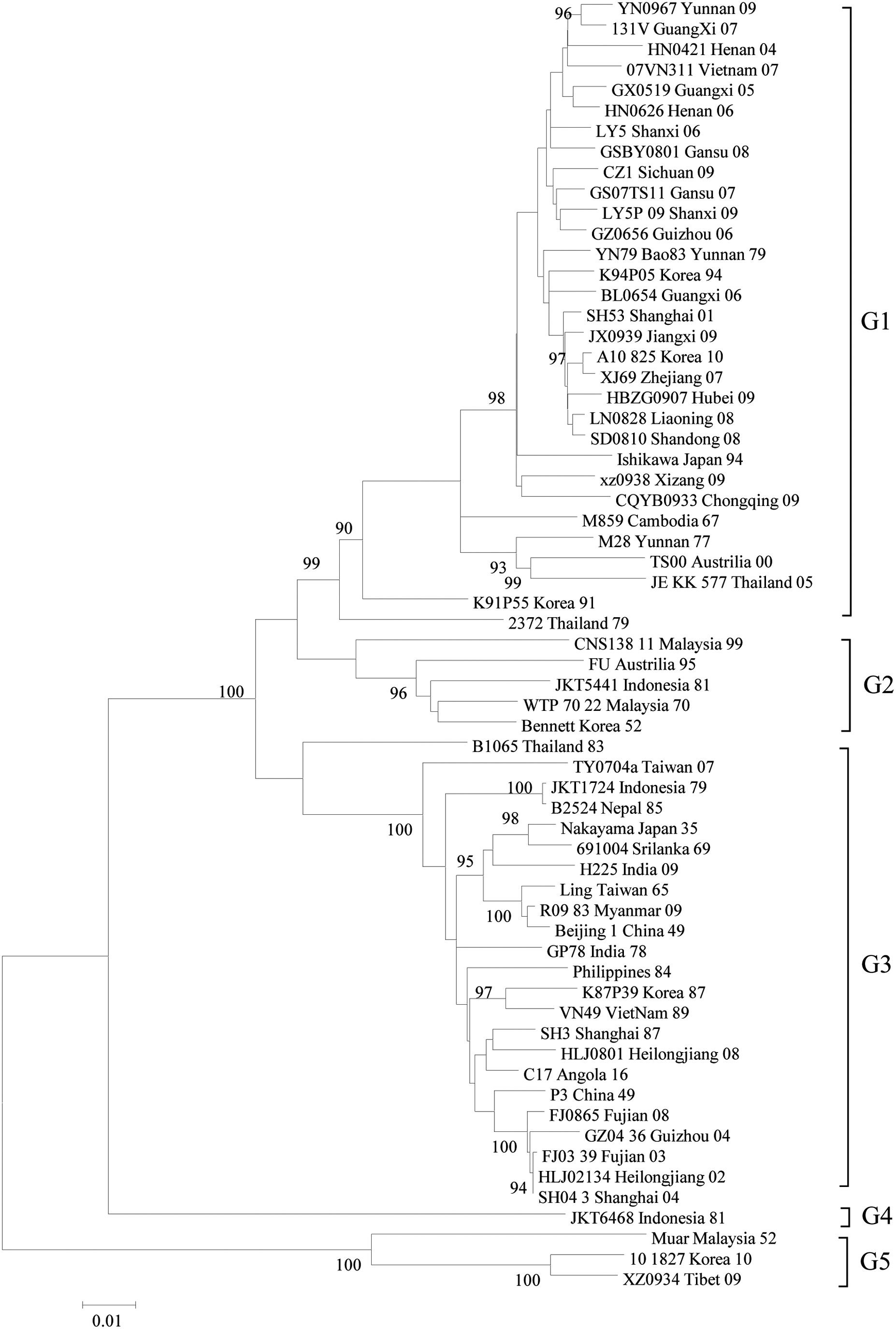
Five genotypes of Japanese encephalitis virus. The detailed information of the temporal and spatial distribution of five genotypes of JEV please see Table 1.
Based on literature review and available sequence data by end of December 19, 2017.
As Table 1 and Figure 1 reveal, the epidemic areas of JEV can be divided into eight regions according to the geographic distribution of genotypes. Among these regions, six (A–F) are traditional epidemic areas of JEV and JE, mainly distributed in Asia and Oceania, while the regions of Italy in Europe (G) and Angola in Africa (H) were previously non-epidemic areas with regard to JEV and JE. Between 1935 and 2017, G1 and G3 JEV had the highest prevalence and widest distribution, while the geographic distributions of G2, G4, and G5 were generally limited to tropical regions in southeastern Asia. JEV G1–G5 had been present in traditional epidemic areas between 1935 and 2000; however, G1, G2, G4, and G5 were prevalent only in tropical southeastern Asia, while G3 appeared in South Asia and East Asia, including most parts of China (G1 was only found in Yunnan Province, southwestern China, adjacent to Thailand and Vietnam and G2 was discovered in South Korea during this period). Since 2000, in contrast, G1 has emerged in almost all traditional epidemic areas, including southeastern Asia, South Asia, and East Asia. Especially in Japan, Korea, and China (including Taiwan), G1 has become the dominant genotype, displacing G3. Furthermore, since 2000, G5 has re-emerged in Tibet, China, and Korea after having disappeared for 50 years.
Geographic Distribution of JEV G1 and Its Relationship with Disease
Geographic distribution of JEV G1
The first strain of JEV G1 was isolated in Cambodia in 1967. It was subsequently isolated in Yunnan Province, China in 1977 (M28 stain) (Wang et al. 2008) and 1979 (YN79-Bao83 strain) (Wang et al. 2007a). These three strains are the earliest G1 isolates. After 1990, G1 was isolated from Australia, and then was continuously present in Thailand, Laos, Vietnam, and Burma, as well as in Central Asia and East Asia, including China, Japan, and South Korea (Table 1). The geographic distribution of JEV G1 has spread to India in South Asia and many provinces of mainland China since 2000. JEV G1 has become the dominant genotype in these areas, displacing G3 (Wang et al. 2007a, Pan et al. 2011, Gao et al. 2013, Schuh et al. 2014).
Molecular genetic analysis of the whole-genome sequences of JEVs isolated from JE patient, animals (pig blood and bats) and more than 20 species of mosquitoes and midges has shown that JEV can be divided into five genotypes. Five population differentiation events occurred during the evolution of JEV, and five independent branches were successively derived, in the order of G5, G4, G3, G2, and G1. Thus, G1 is the youngest JEV species. Moreover, population dynamics analysis revealed that G1 JEV has been dominant in competition with G3 since 1950 (Pan et al. 2011, Gao et al. 2015).
In a previous study, the sequences of E genes from JEV G1 and G3 were obtained to analyze the temporal and spatial distribution of JEV in Asia (Schuh et al. 2014). As mentioned in the introduction, the study showed that JEV G1 was divided into two large clades, GI-a and GI-b. GI-a can be further divided into the Thailand subgroup and Cambodia subgroup, which have been continuously prevalent in the tropical regions of Asia, while GI-b spread northward from Vietnam, through China (including Taiwan), to Japan and Korea in East Asia. Thus, the GI-b clade was the major JEV population (Schuh et al. 2014) that was widespread in Asia since the 1990s, and which has gradually replaced G3 as the dominant genotype in Asia. There were molecular adaptation sequences at residue 15 of the G1 E protein, as well as co-evolution loci (residues 89 to 360 and 129 to 141). The study on JEV replication and temperature sensitivity using chicken and mosquito cells showed that the virulence of the GI-b strain (JE-91) inoculated in mosquito cells (C6/36) was greater than that of G1-a and G3. That is, G1-b had stronger pathogenicity in mosquitoes than G3, which may be one of the reasons that JEV G1 is becoming the dominant genotype in Asia (Schuh et al. 2014).
JEV G1 and viral encephalitis
An outbreak of JE in adults occurred in Yuncheng, Shanxi Province in central China in 2006, where 66 cases were reported, including 19 deaths (fatality rate of 28.8%) during July–August. Children under 7 years old accounted for 13.4% of cases, while adults over 30 years old accounted for 86.6%. In addition, 94.74% of deaths were in the age group of over 50 years. Six of the 13 cerebrospinal fluid (CSF) specimens were PCR-positive for JEV. Ten of the 24 pools of mosquito specimens were PCR-positive for JEV. Nucleotide sequencing suggested that amplification products from specimens of patients and mosquitoes (six patients, five mosquitoes) belonged to JEV G1 and G3. JEV G1 and G3 could be isolated from both the patient and mosquito samples. These results suggested that the epidemic of adult JE cases might be caused by co-infection with G1 and G3 (Wang et al. 2007b).
Five JEV PCR-positive cases were detected from CSF specimens of patients diagnosed with JE in Guizhou Province and Yunnan Province in southwestern China in 2006 and 2008, respectively (Wang et al. 2010). Sequence analysis indicated they belonged to G1. One viral strain (GZ56) was obtained and identified as G1 through whole-genome sequencing and analysis. The G1 strain isolated from a CSF specimen had high homology with that isolated from mosquitoes collected in the same area. In addition, 31 specimens of CSF were collected from JE patients in Yunnan Province in southwestern China and Shanxi Province in central China in 2009, and three viral isolates that could cause cytopathogenic effects (CPE) were obtained after tissue culture (Zhang et al. 2009). Whole-genome sequence analysis demonstrated that all three isolates were JEV G1. These strains were isolated from a 4-year-old girl in Yunnan Province, and a 2-year-old boy and an 82-year-old female patient in Shanxi Province.
From September to November of 2009, a large number of viral encephalitis cases were reported in the Gorakhpur region of India. JEV G1 was identified in 27 CSF specimens, indicating that JEV G1 had become the dominant genotype causing JE in this region (Fulmali et al. 2011).
Geographic Distribution of JEV G2 and Its Relationship with Disease
Geographic distribution of JEV G2
The WTP-70-22 strain isolated from Malaysia in 1970 is generally believed to be the first observed JEV G2 strain. Subsequently, the JKT1749 and JKT5441 strains isolated in Indonesia in 1979 and 1981, respectively, the B1065 strain from southern Thailand in 1983, and the FU strain isolated in Papua New Guinea and northern Australia in 1995, as well as the CNS138-11 strain isolated in Malaysia in 1999, all fell into JEV G2 (Table 1). It seems that the geographic distribution of JEV G2 is limited to the region between northern Australia (20°S) and southern Thailand (10°N), and has a history of only about 30 years due to the timing of its isolation in 1970 to 1999 (Williams et al. 2000, Schuh et al. 2010, 2011).
Genomic sequencing and analysis of 16 JEVs isolated from patients with viral encephalitis in South Korea and Japan in the 1930s–1970s found 15 strains belonging to JEV G3, while 1 strain (in Korea; Bennett strain 1951) was JEV G2 (Schuh et al. 2011). Bennett strain was isolated from an infected individual during a JE outbreak among 300 American soldiers. The sick individual, like other American soldiers, had no history of being bitten by mosquitoes before reaching Korea. Therefore, it can be assumed that the soldier was infected with JEV in Korea, and the disease was not an imported case. These results may potentially indicate that JEV G2 was prevalent in Korea and southeastern Asia in the 1950s. The isolation of the Bennett strain shifted the isolation time of the JEV G2 genotype 20 years earlier than previously thought. At the same time, it is also the finding of endemic JEV G2 north of Thailand.
JEV G2 and viral encephalitis
From March to April 1995, three JE patients were identified on Badu Island in the Torres Strait, northern Australia, two of whom died. These patients were initially thought to be infected with MVE virus, but JEV infection was later confirmed. These cases represent the first identification of JEV infection in northern Australia, which is about 2000–3000 kilometers away from the nearest JE epidemic area. Subsequent investigations revealed that positive JE infection was detected in 55 patients dwelling on four islands around the epidemic area as well as 90 pigs on nine islands. In total, 10 JEV G2 strains were isolated in this outbreak. Among them, two strains came from patients with sub-clinical manifestations and the other eight were isolated from local Culex pipiens specimens (Cx. annulirostris). These results revealed that there was a natural circulation of JEV in the area (Hanna et al. 1996). To prevent further spread of the epidemic, the local population was vaccinated with an inactivated JE vaccine in December 1995 and January 1996.
Torres Strait was attacked by JEV again in 1998. A child who was not vaccinated with JE vaccine was infected. In addition, a seroepidemiological survey in pigs demonstrated further expansion of the JEV epidemic area. A total of 45 JEV G2 strains were isolated from Badu Island, of which 1 strain was obtained from a pig specimen and another 44 strains were originated from mosquito specimens (43 pools of Cx. annulirostris and one pool of Ochlerotatus vigilax). JEV was still active in this area in 2000 and 2001, and the JEVs were isolated from three sentinel pig serum specimens and a group of Cx. gelidus. However, JE cases were not found in the local population (Hanna et al. 1996, Mackenzie et al. 2002).
The JEVs isolated from Australia in 1995 and 1998 belonged to JEV G2, which belongs to the same evolutionary branch as the JEV G2 isolates from Malaysia in 1970 and Thailand in 1983, suggesting that Australia's G2 may have originated from Malaysia or Thailand (Hanna et al. 1996, Williams et al. 2000, Mackenzie et al. 2002). However, the JEV isolated from Australian pigs and mosquitoes in 2000 belonged to G1, demonstrating the prevalence of two JEV genotypes in northern Australia between 1995 and 2000.
As mentioned above, JEV G2 (Bennett strain) was isolated from Korean encephalitis patient specimens, suggesting that JEV G2 caused JE case in East Asia in the 1950s, although there was significant circulation of JEV G3 during that time and most patients were infected with G3 (Schuh et al. 2010, 2011).
Geographic Distribution of JEV G3 and Its Relationship with Disease
Geographic distribution of JEV G3
As shown in Table 1, JEVs isolated from 20 countries or regions, including Japan, South Korea, Thailand, Vietnam, Malaysia, Indonesia, India, Sri Lanka, Philippines, Nepal, and 10 provinces or regions of China between 1935 and 1990 belonged to G3. These areas cover the majority of the 24 JE epidemic countries or regions reported by the WHO, indicating that JEV G3 was the dominant genotype during this period.
The first JEV G3 strain, which was also the earliest JEV strain (Nakayama), was obtained from brain tissue of JE patients in Tokyo, Japan in 1935, and is the prototype strain of JEV (Lindenbach et al. 2007, Halstead and Jacobson 2008). Since then, JEV was isolated from the brain tissue of a patient with viral encephalitis collected in Beijing, China in 1943 (the virus was not preserved). Subsequently, P3 was isolated from brain tissue specimens of patients in Beijing in 1949. All of these isolates belong to JEV G3 (Li et al. 2004, Zheng et al. 2012). As noted above, molecular genetic analysis of 16 JEV strains isolated in Japan and Korea in the 1930s–1970s showed that 15 strains were JEV G3 and 1 was JEV G2 (Schuh et al. 2010, 2011). These results indicate that JEV G3 was the main genotype in Japan, Korea, and China in the 1930s–1970s.
However, since the 1990s, especially since entering the 21st century, JEV G1 has gradually emerged in Japan, South Korea, and mainland China where previously JEV G3 circulated. Moreover, the geographic distribution of JEV G1 has gradually expanded, while the distribution area of G3 has been reduced, thus it appears that JEV G1 is replacing JEV G3 in Asia (Pan et al. 2011, Schuh et al. 2014). Although JEV G3 was gradually replaced by G1 in the traditional endemic areas, G3 has spontaneously appeared outside of the traditional endemic areas for JEV and JE, specifically in Europe and Africa. For example, JEV G3 was detected in mosquito and bird specimens (Platonov et al. 2012, Ravanini et al. 2012) in Europe, and one local JE case was reported in Africa (Simon-Loriere et al. 2017).
JEV G3 strains (NS5 gene amplification positive) were detected in a pool of C. pipiens mosquito specimens collected in northeastern Italy in the summer of 2010. Unfortunately, viral isolation and amplification of other segments of JEV were all negative (Ravanini et al. 2012). Positive JEV gene amplification was subsequently detected in bird specimens collected in Tuscany, where JEV-positive mosquitoes had been collected (GenBank accession numbers: AF501311–AF501315). Phylogenetic analysis demonstrated that this strain belonged to JEV G3, and had the closest genetic relationship with the Nakayama strain first isolated in Japan in 1935. Epidemiological analysis found that no JE patients existed in the area where the bird specimens were collected. Therefore, it is believed that there was a limited epidemic cycle of JEV between birds and mosquitoes in southern Italy (Platonov et al. 2012). The reason that JEV did not spread on a larger scale despite the natural cycle of JEV was established may be the lack of pigs, the primary hosts of JEV (Platonov et al. 2012). In any case, detection of JEV from both birds and mosquitoes suggests that JEV has spread to Europe, at least Italy, from the traditional JE epidemic areas in Asia.
JEV G3 and viral encephalitis
Historical JE outbreaks caused by JEV G3
JEV G3 caused a series of JE outbreaks throughout Asia in the 1930s to 1970s. JE was first documented in Japan in 1871. JE broke out in Tokyo in 1924, resulting in 6000 JE cases, of which 60% resulted in mortality (Lindenbach et al. 2007, Halstead and Jacobson 2008). JEV (Nakayama strain) was first isolated in Japan in 1935. Since then, JE vaccine was developed, and the incidence of JE in Japan has declined sharply due to widespread inoculation with the JE vaccine. Currently, only a few JE cases are reported annually (Lindenbach et al. 2007, Halstead and Jacobson 2008). In South Korea, about 4000 cases of JE were reported in 1966. JE vaccination program was initiated in South Korea at the beginning of the 1970s and JE vaccine was included in the Expanded Program on Immunization (EPI) in 1985. As a result, the incidence of JE in South Korea has been significantly reduced (Lindenbach et al. 2007, Halstead and Jacobson 2008).
Many JE epidemics have been recorded in Chinese history. The wide range, high incidence, and heavy disease burden of these epidemics were unusual compared to those in other regions (Zheng et al. 2012). JE became a notifiable infectious disease since 1951 in China. More than 1.4 million JE cases were reported during the period of 1963–1975, accounting for 60% of all cases between 1950 and 2013. The incidence of JE in China was between 8.32 and 20.92 per 100,000 in this period. Nearly 150,000 and 170,000 JE cases were reported in 1966 and 1971, with morbidity up to 20.58/100,000 and 20.92/100,000, respectively, without JE vaccine intervention (Gao et al. 2014, Li et al. 2014b). Since then, China has gradually popularized JE vaccines (inactivated vaccine and attenuated live vaccine) through its own national development and production system, controlling the infection rate and incidence of JE significantly and achieving remarkable results. The incidence of JE was reduced from 20.92/100,000 in 1971 (about 170,000 reported cases) to 0.12/100,000 in 2011 (1625 JE cases reported).
Local case of JE in Angola, Africa
A 19-year-old male patient was hospitalized due to five consecutive days of fever, headache, and jaundice during a yellow fever epidemic in Angola, Africa in March, 2016. A positive PCR result for yellow fever virus was obtained from the blood of the patient. High throughput sequencing was performed using cDNA samples prepared during detection of yellow fever in the hospital. Surprisingly, sequencing results suggested that there was not only yellow fever virus (GenBank accession No. KX982182), but also the sequence of JEV (GenBank accession No. KX945367) in this specimen. Phylogenetic analysis demonstrated that the JEV detected from this specimen was G3. The patient was considered locally infected, as he worked in the area and did not have a history of traveling abroad. This represents the first reported JE case in Africa, outside of the traditional epidemic areas in Asia and Oceania. These results also suggest that JEV has a potentially natural cycle in Africa, or that Africa is likely to become a new focus of JEV (Simon-Loriere et al. 2017).
Many viral encephalitis epidemics are still caused by JEV G3
A huge outbreak of JE occurred in the Gorakhpur region of Uttar Pradesh, India in July–November, 2005, with 5737 persons infected and 1344 fatalities. Seven strains of JEV were isolated from patient specimens, and the molecular genetic analysis of the viral genome showed that all belonged to JEV G3 and formed a separate evolutionary cluster. This outbreak is the most severe JE epidemic in India in the last three centuries (Manmohan et al. 2006). JEV G3 is the dominant genotype in the Gorakhpur region of northern India (Saxena et al. 2009).
Geographic Distribution of JEV G4 and Its Relationship with Disease
As of 2017, JEV G4 has only been isolated from mosquito specimens collected in Indonesia, and its isolation time is limited to 1980–1981 (Schuh et al. 2013). There has been no JEV G4 or viral encephalitis caused by it reported in other areas. JEV G4 has the most limited distribution among the five genotypes.
Geographic Distribution of JEV G5 and Its Relationship with Disease
The first isolated strain of JEV G5
Four cases of viral encephalitis were diagnosed in Malaya (one case) and Singapore (three cases) in the summer of 1952. All patients had clinical manifestations including high fever, vomiting, headache, disturbed consciousness, neck stiffness, and deep coma. Soon, all patients died due to respiratory failure and other symptoms. Four viral isolates were obtained from brain tissue specimens, which were identified as JE through a neutralization test with the Nakayama strain (Hale et al. 1952). Subsequent viral molecular genetic analysis demonstrated that the JEV (Muar strain) isolated from a 19-year-old male patient in Malaya in 1952 was a JEV G5 strain (Hasegawa et al. 1994, Solomon et al. 2003). This is also the first JEV G5 strain.
The second JEV G5 strain isolated
A viral isolate (XZ0934) was obtained from a Cx. tritaeniorhynchus specimen collected in Tibet, China in 2009. The virus caused CPE in BHK-21 cells and C6/36 cells (72 h). Whole-genome sequence analysis showed that the full-length genome of XZ0934 was 10,983 nt, consisting of a single ORF. Phylogenetic trees were constructed separately based on the C, PrM, M, and E genes, and the results showed that XZ0934 is in the same evolutionary branch as JEV G5 (Muar strain), suggesting that the XZ0934 virus was a novel G5 isolate (Li et al. 2011, 2014a). This is the second G5 isolated in the world, indicating that JEV G5 re-emerged after being silent for 60 years (1951–2009).
JEV G5 detected in mosquito specimens in East Asian regions
Through an arbovirus investigation performed in Korea in 2008–2010, positive PCR results for JEV were detected in 64 pools of mosquito specimens, among which 18 pools contained Cx. tritaeniorhynchus specimens, and one pool was made up of Cx. bitaeniorhynchus specimens. Sequencing and phylogenetic analysis of the E gene revealed that the positive result from Cx. bitaeniorhynchus was JEV G5. This is the first record of JEV G5 in Korea, as well in the East Asian region, after its discovery in Tibet, China in 2009. Since then, JEV G5 has been discovered in many regions of South Korea (Takhampunya et al. 2011, Kim et al. 2015).
Challenges
Numerous studies have verified that JEV has five genotypes (Pan et al. 2011, Gao et al. 2015). Each genotype exhibits unique geographical distribution characteristics, but all are capable of causing disease epidemics. The geographic distribution of these genotypes is not fixed. For example, JEV G1 is becoming more and more widespread (Wang et al. 2007a, Pan et al. 2011, Gao et al. 2013, Schuh et al. 2014). G3 is no longer confined to the traditional endemic areas of JEV, but also appears in non-traditional regions (Platonov et al. 2012, Ravanini et al. 2012, Simon-Loriere et al. 2017). G5 re-emerged after disappearing for more than 50 years (Li et al. 2011, 2014a, Takhampunya et al. 2011, Kim et al. 2015). These changes are certain to create new challenges. In recent years, fewer cases caused by JEV G3 have been diagnosed in traditional epidemic areas (Nga et al. 2004, Wang et al. 2007a, Nitatpattana et al. 2008, Pan et al. 2011, Schuh et al. 2014). However, JEV G3 has appeared in new regions such as Africa and Europe, indicating that the ecological activity of JEV G3 may affect future global disease burden. The disease burden brought by the re-emerged G5 needed careful evaluation. Furthermore, whether JEV G5 will further spread through the traditional JE epidemic areas via transmission paths similar to those of JEV G1 requires continuous monitoring. The number of JE cases has been maintained below 10 annually in Korea and Japan before 2000. However, 129 JE cases were reported in Korea in 2010–2015 (Sunwoo et al. 2016), primarily in elderly patients. Whether the re-emergence of JE in Korea is related to JEV G5 requires further study. JE is a vaccine-preventable disease, but recent research suggests that current JE vaccines (both inactivated and attenuated) do not provide complete protection against JEV G5 attack (Cao et al. 2016). Whether or not a new vaccine is needed to deal with this challenge is still under debate.
In conclusion, JEV remains an important encephalitis virus that threatens human health. Any time a new genotype occurs in the traditional epidemic areas or JEV appears in non-traditional epidemic areas, attention is warranted. We must continuously strengthen the monitoring and detection efforts for JEV in traditional epidemic areas. It is also necessary to detect occurrences of JEV in non-traditional epidemic areas and minimize the public health burden associated with this virus.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81501757 and 81301479), the National key research and development project (2016YFC1201904, 2016YFC1200905, 2017YFC1200503), the Development Grants of State Key Laboratory of Infectious Disease Prevention and Control (2014SKLID103, 2015SKLID505), Open Research Fund Program of CAS Key Laboratory of Special Pathogens and Biosafety, Wuhan Institute of Virology, Funded by Open Research Fund Program of Wuhan National Bio-Safety Level 4 Lab of CAS (2017SPCAS003). The founders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author Disclosure Statement
No competing financial interests exist.