
Abstract
The Rocky Mountain wood tick, Dermacentor andersoni, has long been known to transmit human pathogens. Within the Bitterroot Valley, Ravalli County, Montana, these agents include Rickettsia rickettsii, Francisella tularensis, and Colorado tick fever virus (CTFV). Found in the western United States where wood ticks occur, CTFV causes a biphasic, febrile illness in humans and persists in enzootic cycles involving the ticks and small mammals. CTFV belongs to the genus Coltivirus, family Reoviridae, whose genome consists of 12 double-stranded RNA segments. Previous studies revealed the presence of CTFV-infected ticks and rodents in select locations within the valley in the 1960s and 1970s, using animal and cell culture methods for detection. We aimed to determine the range and prevalence of the virus in adult questing ticks throughout the valley using molecular tools and to examine the genomic variation between virus strains. Adult D. andersoni ticks were collected during 2002–2003 and 2009–2013. RNA extractions and reverse transcription-polymerase chain reaction were performed on 921 ticks, of which 61 ticks were positive for CTFV, resulting in a 6.6% prevalence of infection. Four genetic loci, one from each of the segments 9, 10, 11, and 12, within the viral genome were sequenced. Reassortment was detected between CTFV sequence strains within the valley. This study confirmed the prevalence of CTFV in D. andersoni ticks within the Bitterroot Valley, which has remained at levels found in the 1950s and 60s. Additional CTFV sequences were obtained and evidence of reassortment was observed between strains within the valley.
Introduction
The Rocky Mountain wood tick, Dermacentor andersoni, found in the western United States and Canada is known to harbor a number of human pathogens (Cooley 1932). Among these are Rickettsia rickettsii, the agent of Rocky Mountain spotted fever; Francisella tularensis, the agent of tularemia; and Colorado tick fever virus (CTFV) (Cooley 1932). CTFV was identified in 1945 as the etiological agent of a disease that, by 1930, had already received the name CTF due to the strong correlation between human disease and exposure to the Rocky Mountain wood tick in the state of Colorado (Topping et al. 1940).
CTFV (genus Coltivirus, family Reoviridae) is a 12-segmented double-stranded RNA virus with a total combined genome length of 29,174 bp (Attoui et al. 2000). Little is known of the proteins encoded by each segment. Some putative protein predictions have been made based on homology to other viral proteins (Attoui et al. 2000). The virus persists in enzootic cycles involving rodents and ticks, and its distribution overlaps the distribution of the tick vector. Humans are accidental hosts that become infected when an infected adult or nymph D. andersoni feeds on them. CTF often presents with a high fever, severe myalgia, and headache, but signs and symptoms can vary (Emmons 1988). The fever is usually biphasic, with a second fever appearing 2–3 days following the disappearance of the first (Klasco 2002).
The Rocky Mountain wood tick is a three-host tick with a 2–3-year life cycle. Larvae emerge in the late spring/early summer and engorge on small mammals such as chipmunks, ground squirrels, and deer mice before dropping off and molting to a nymph. The nymphs feed again on small mammals in the late summer and molt to adults or overwinter unfed. The next spring, unfed nymphs and adults emerge and feed. Adults quest on grass or shrubs waiting to attach to passing large mammals, such as deer, elk, or livestock. If an adult is unable to feed, it may die or overwinter again and reemerge the next season to find a host. Mating occurs on the host. Engorged females drop off, lay eggs, and die (Cooley 1932). Ticks acquire the virus as larvae or nymphs from infected rodents and remain infected for life, but the virus is not transovarially transmitted (Eklund et al. 1959).
The Bitterroot Valley in Ravalli County of western Montana is endemic for wood ticks infected with CTFV (Cooley 1932). A number of studies done in the valley in the late 1950s and early 1960s found a variety of rodent species important for the maintenance of the viral infection cycle. A study in the Lost Horse drainage, about nine miles southwest of Hamilton, Montana, found the primary CTFV reservoir to be the golden mantled ground squirrel, Callospermophilus lateralis. At Mill Creek, about five miles northwest of Hamilton, Peromyscus maniculatus, the deer mouse, and Neotoma cinerea, the bushy-tailed wood rat, were the principal reservoirs (Eklund et al. 1959, Burgdorfer 1960, Clark et al. 1970). A variety of hosts maintain the virus in a given area, and the primary mammal species involved varies by location. A range in the prevalence of infection of the virus within D. andersoni ticks and rodents was also noted at these locations.
The purpose of this study was to reassess the prevalence of CTFV within the tick population. This information will increase our understanding of the local presence of the virus as well as strengthen our general knowledge of the ecology and molecular composition of CTFV.
Materials and Methods
Tick collection
Ticks were collected by the authors, local hikers, and Bitterroot National Forest (BNF) employees. Most ticks were collected during 2009–2013, although a small number of ticks collected between 2002 and 2003 were also tested. Forest Service employees were given tubes to collect ticks when working in the field. Collection tubes were constructed by cutting an “X” slit in the cap of a 15 mL tube (Corning, Corning, NY) and pushing a 1.5 mL screw cap tube through the slit until it fit snuggly in the cap. The end of the small tube was removed, allowing for ticks to be placed in through the smaller tube, while the “collar” of the smaller tube prevented the already collected ticks from climbing out. Ticks found on a person's clothes or body were placed into tubes labeled with the date and location. Flagging for ticks at locations known to contain CTFV-infected ticks from previous studies was also done. Ticks were either immediately crushed for RNA extraction or stored at −80°C until processing.
RNA extraction
RNA extractions were performed in an RNase-free area with RNase-free products using the RNeasy Mini Kit (QIAGEN, Germantown, MD). Whole ticks were placed individually into 1.5 mL snap-cap tubes with a disposable pestle (Fisher Scientific, Pittsburgh, PA) and flash-frozen in liquid nitrogen. Ticks were crushed into a fine powder, suspended in 600 μL of RLT buffer, and run through a QIAshredder column (QIAGEN) to homogenize the sample. Each sample extraction was performed following the manufacturer's instructions for animal tissues, including an on-column DNase digestion using RNase-free DNase I (QIAGEN) and eluted in 50 μL nuclease-free water.
Screening samples by reverse transcription-polymerase chain reaction for CTFV
Samples were first tested for infection by targeting genomic segment 12 of the virus. Denaturation of the ds-RNA was performed by diluting 3.96 μL of sample in 2.04 μL of a 1.4% formamide solution, heating to 95°C for 5 min, and immediately putting the samples on ice. Reverse transcription-polymerase chain reaction (RT-PCR) was performed using the Access RT-PCR System (Promega, Madison, WI) in a total reaction volume of 20 μL. Final concentrations of reagents were as follows: 1 μM of each primer CTFVsg12F and CTFVsg12R (Table 1), 200 μM dNTP, 1 mM MgSO4, 0.1 unit AMV reverse transcriptase, 0.1 unit Tfl RNA polymerase, and 1.52 μL of the formamide-RNA extraction dilution. Reactions were heated in a thermal cycler to 45°C for 45 min followed by 95°C for 5 min and 35 cycles of 94°C for 30 s, 55°C for 1 min, and 68°C for 2 min before a final extension at 68°C for 12 min. Portions of three additional genomic segments (9, 10, and 11) were amplified for RNA samples positive for CTFV from the initial screen, using the primers listed in bold type in Table 1.
Primers Used in This Study
Primers in bold were used for the initial amplification; all primers were used for sequencing.
CTFV, Colorado tick fever virus.
Sequencing and analysis
All segments were Sanger sequenced using primers listed in Table 1 and analyzed using Sequencher 5.4.1 DNA sequence analysis software (Gene Codes Corp, Ann Arbor, MI). Evolutionary analyses were conducted using MEGA5 (
GIS map generation
Maps were created using ESRI ArcGIS 10.4.1 for Desktop. All ticks were collected by hikers and forest service workers who did not have GPS instruments during collection. The locations were given as local place names. For trails, trailhead coordinates were used. Nontrail landmarks were searched using TopoQuest (
Results
Ticks were collected over an 11-year period, 2002–2013, from several locations throughout the Bitterroot Valley and BNF (Fig. 1). The collection range included both the Bitterroot and Sapphire Mountains and private land in the Bitterroot Valley. Tick collection efforts were focused in popular hiking and recreation areas (coordinates in Supplementary Table S1). Most ticks were collected from Mill Creek (n = 242), Lake Como (n = 213), Blodgett Canyon (n = 80), and the Sawmill drainage east of Stevensville (n = 103). Of the 921 adult D. andersoni tested, 61 ticks (21 of 572 males, 40 of 349 females) had RT-PCR evidence of CTFV infection using the segment 12 primer set, resulting in a 6.6% prevalence of infection (Tables 2 and 3). Throughout the valley, prevalence of infected ticks varied by location from 0% to 20%. The highest prevalence with 20.3% ticks infected was found in Blodgett Canyon.
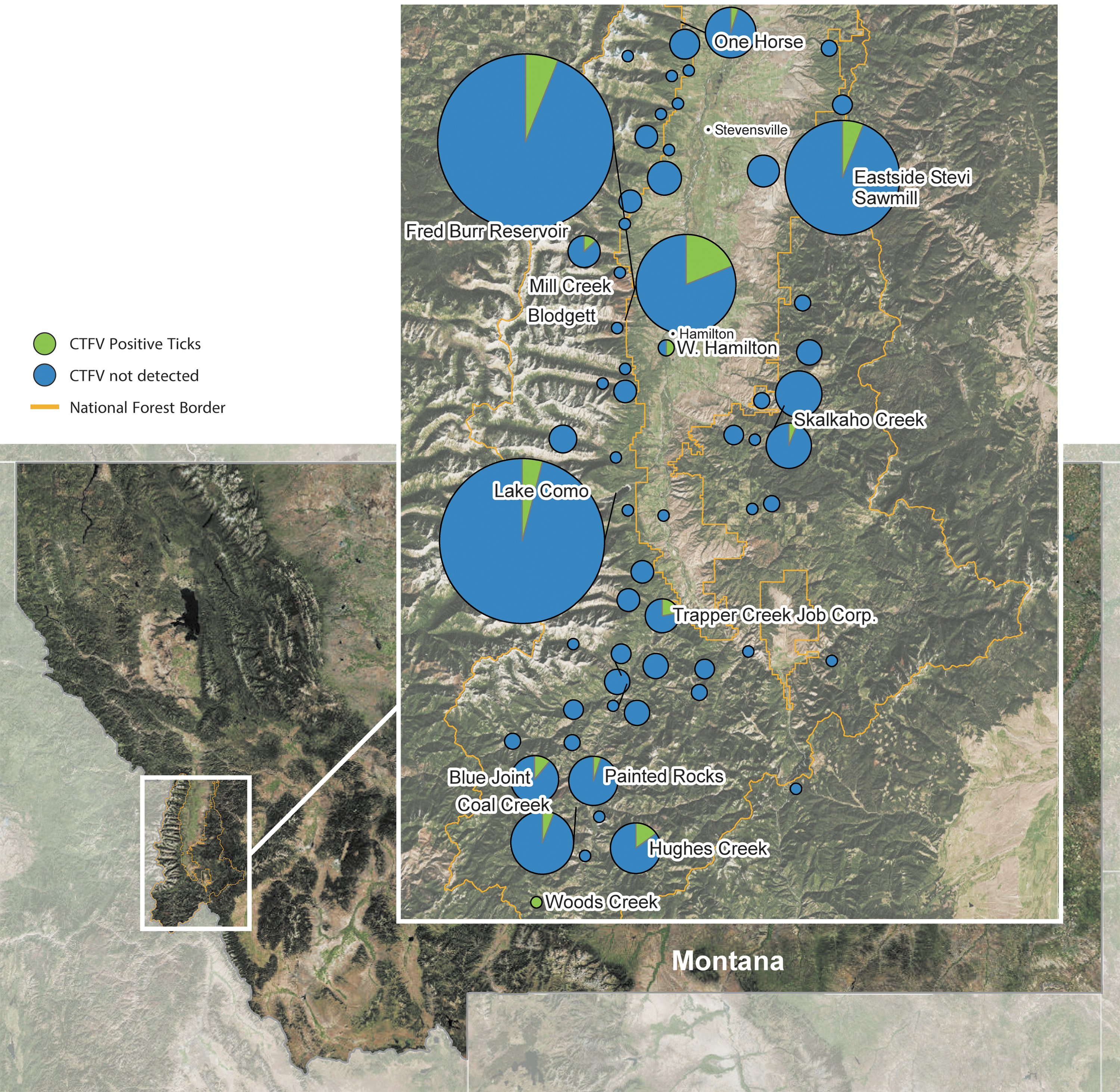
A map of the geographic area in which Dermacentor andersoni ticks were collected. The relative size of the pie graph is proportional to the number of ticks collected at that location. The number of collected ticks ranged from 1 to 243. The green portion of the circle represents the number of ticks positive for CTFV collected at that location. The map was created using ESRI ArcGIS 10.4.1 for Desktop. CTFV, Colorado tick fever virus.
The 27 Strains with Sequence for All Segments 9–12
Data on All Colorado Tick Fever Virus Strains Sequenced
Cr., Creek.
Sequencing was performed for fragments of four viral segments (9–12). Sequences were submitted to GenBank under the following accession numbers: KY454213-KY454264 (segment 9), KY454265-KY454293 (segment 10), KY454294-KY454341 (segment 11), and KY454342-KY454402 (segment 12). Sequence for all four segments was obtained for 27 of the 61 CTFV-positive RNA extractions, herein referred to as sequence strains (Table 2). Total sequence length obtained for each segment varied by segment and strain (Table 3). Sequence lengths used for DNA alignment were 1354 bases for segment 9, 582 bases for segment 10, 249 bases for segment 11, and 527 bases for segment 12.
Percent nucleotide identity among the strains was above 86% in all the segments: 88.33–99.92% (segment 9), 86.04–99.66% (segment 10), 88.35–100% (segment 11), and 95.87–100% (segment 12). Sequence similarities among the strains were highest in segment 12 comparisons and lowest in segment 10 comparisons. Interestingly, while the nucleotide sequences for segment 11 had a high degree of variability, the amino acid sequences were ≥97% identical (including Florio, the reference strain from a human case from Colorado in 1943), suggesting a high selective pressure on conservation of the amino acid sequence (data not shown).
Phylogenetic and sequence analysis of partial sequences of the four segments of the 27 CTFV strains displayed a possible occurrence of segment reassortment (Figs. 2–5). The sequence relatedness of the strains was different between the segments. Three clades comprised the segment 9 tree (Fig. 2). Twenty-two of the strains clustered into clade I. Clade II had one strain, Da389, and clade III contained four strains. Segment 10 comprised five clades. The strains from segment 9 clade II split; four aligned in the segment 10 clade I, while one, Da420, aligned with clade V (Fig. 3). The sequence similarity clustered the strains for segment 11 into two clades (Fig. 4). All sequences for segment 12 were within 95% nucleotide similarity and were assigned one clade (Fig. 5).
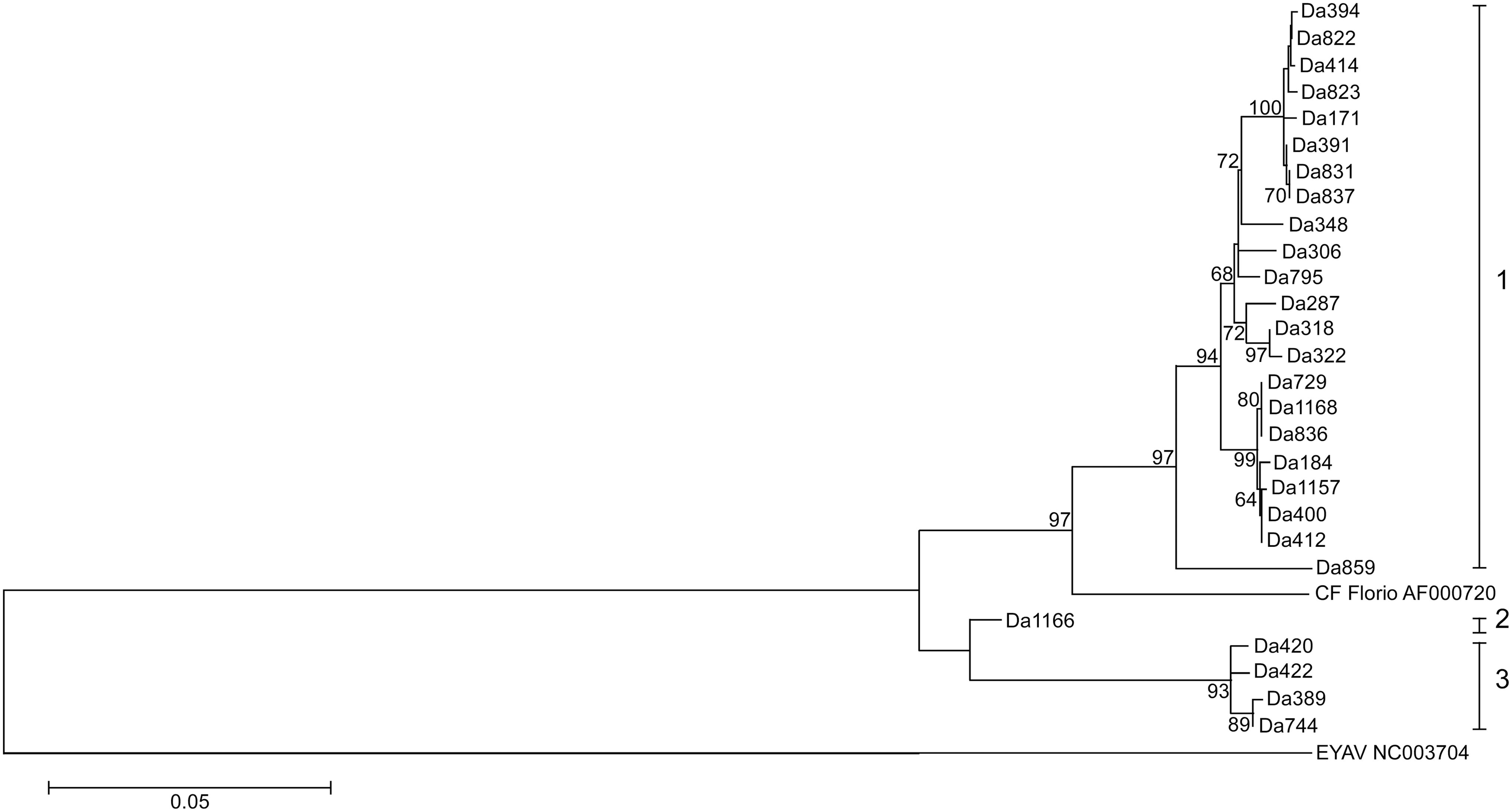
Phylogenetic tree of Bitterroot CTFV tick variants of segment 9 (1353 bases). The tree was inferred using the maximum-likelihood method based on the Tamura-Nei model. Bootstrap percentages (1000 replicates) above 60% are shown next to the branches. The evolutionary distances (branch lengths) were computed using the maximum composite likelihood approach and units are the number of base substitutions per site. EYAV was used as an outgroup. Evolutionary analyses were conducted in MEGA5.
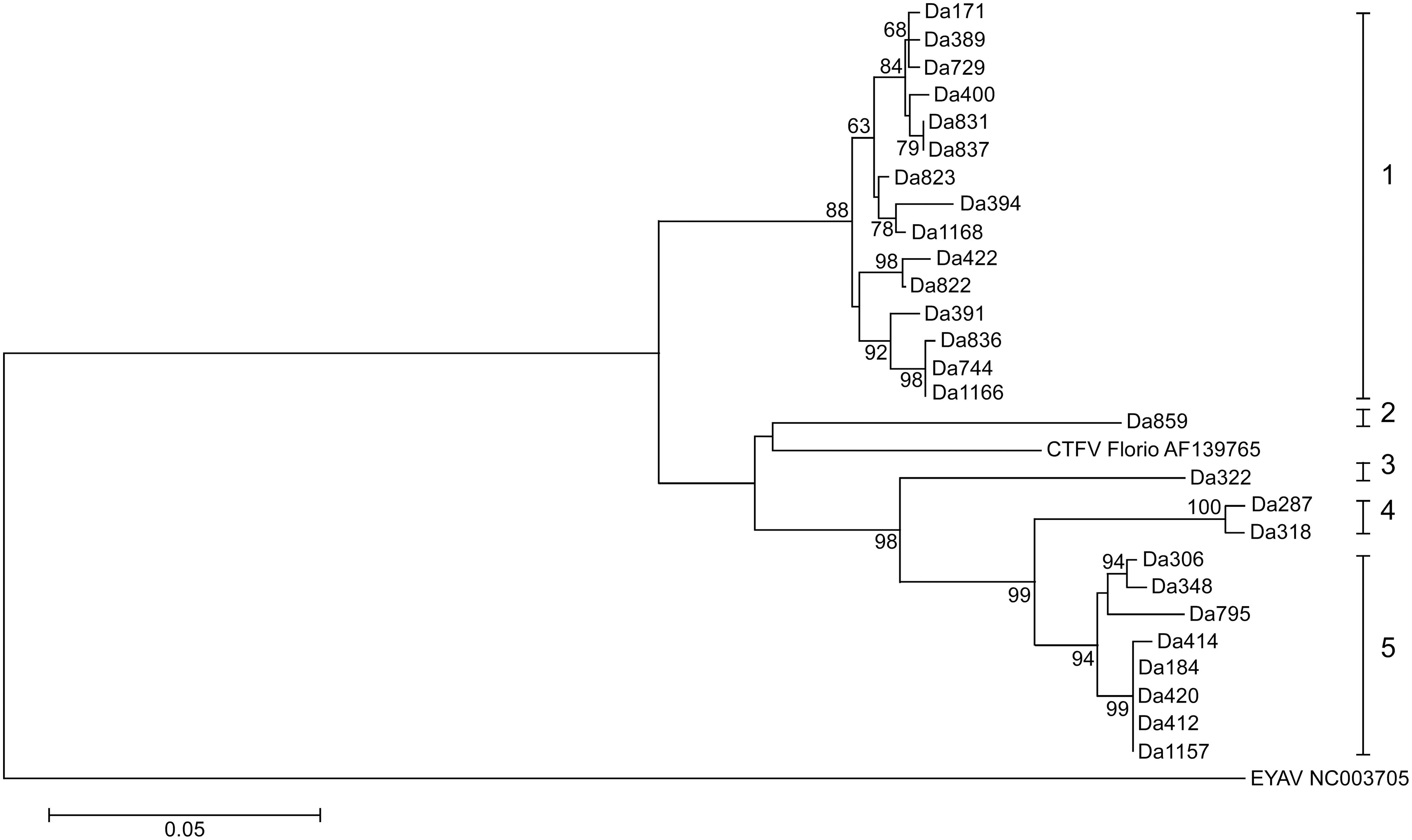
Phylogenetic tree of Bitterroot CTFV tick variants of segment 10 (582 bases). The tree was inferred using the maximum-likelihood method based on the Tamura-Nei model. Bootstrap percentages (1000 replicates) above 60% are shown next to the branches. The evolutionary distances (branch lengths) were computed using the maximum composite likelihood approach and units are the number of base substitutions per site. EYAV was used as an outgroup. Evolutionary analyses were conducted in MEGA5.
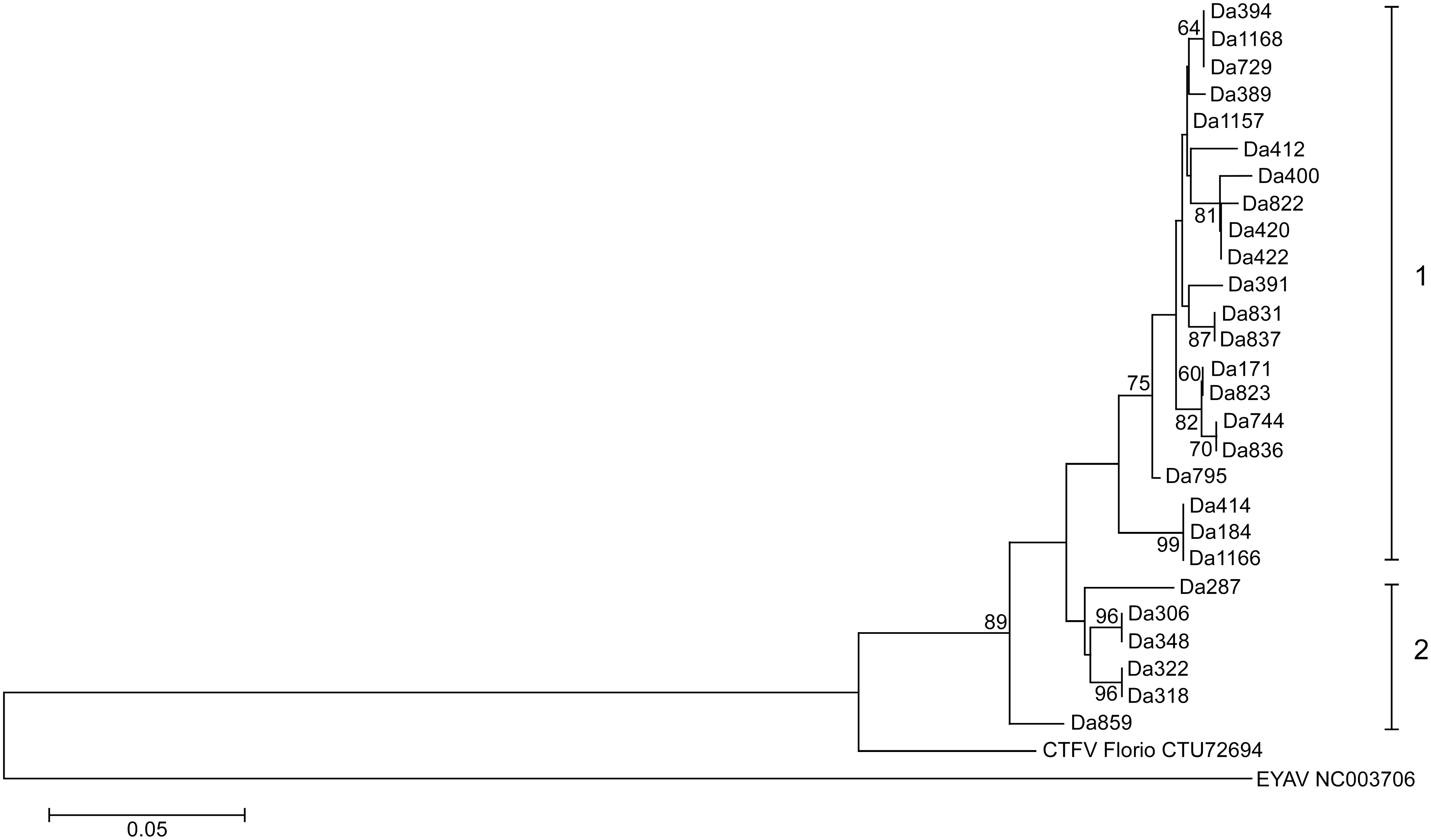
Phylogenetic tree of Bitterroot CTFV tick variants of segment 11 (249 bases). The tree was inferred using the maximum-likelihood method based on the Tamura-Nei model. Bootstrap percentages (1000 replicates) above 60% are shown next to the branches. The evolutionary distances (branch lengths) were computed using the maximum composite likelihood approach and units are the number of base substitutions per site. EYAV was used as an outgroup. Evolutionary analyses were conducted in MEGA5.
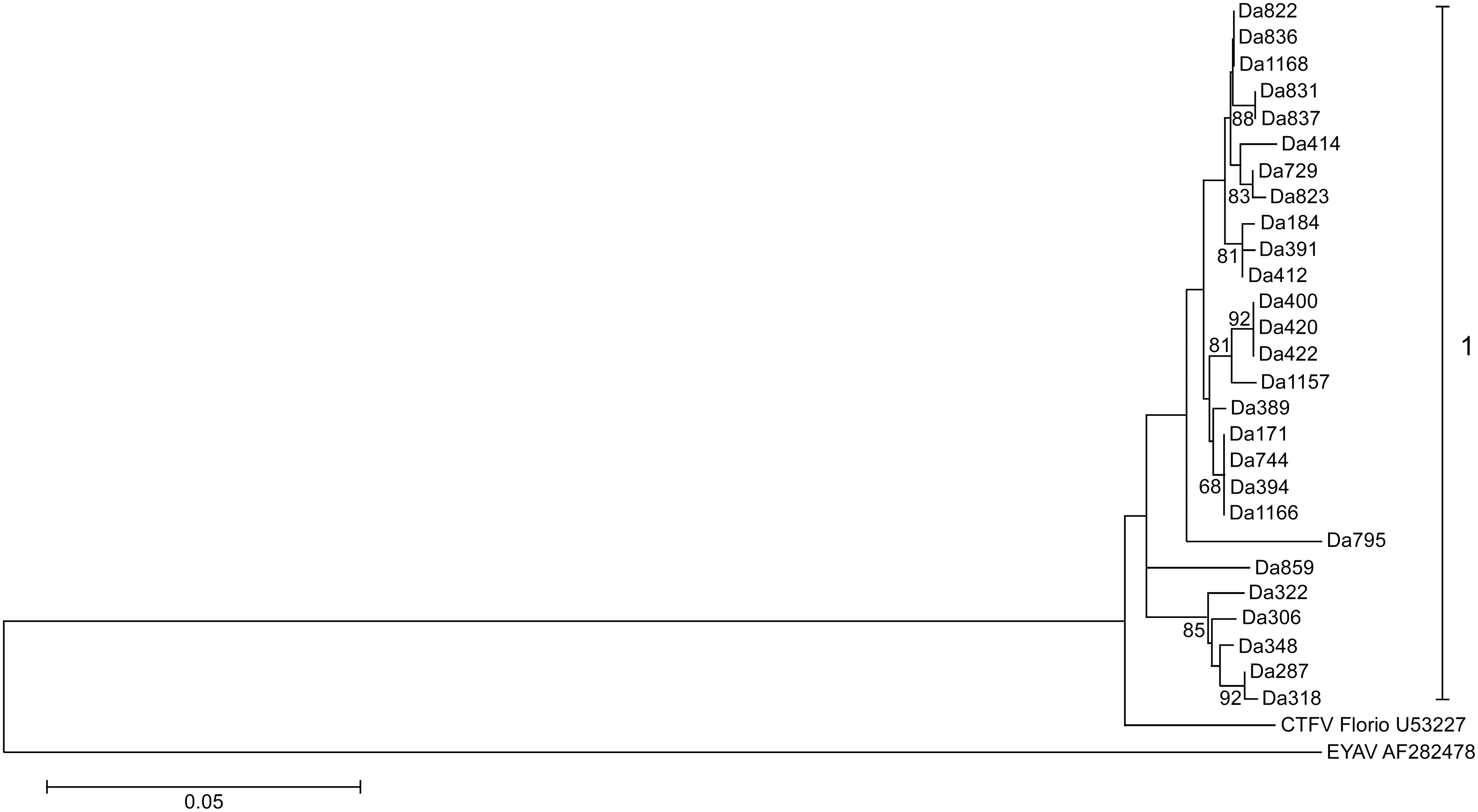
Phylogenetic tree of Bitterroot CTFV tick variants of segment 12 (527 bases). The tree was inferred using the maximum-likelihood method based on the Tamura-Nei model. Bootstrap percentages (1000 replicates) above 60% are shown next to the branches. The evolutionary distances (branch lengths) were computed using the maximum composite likelihood approach and units are the number of base substitutions per site. EYAV was used as an outgroup. Evolutionary analyses were conducted in MEGA5.
The 27 sequence strains that were included in phylogenetic analysis were assigned clade numbers according to each phylogenetic tree (Table 2). Strains were then placed into groups by clade assignment pattern. Nine groups, labeled A to I, were formed, ranging from 1 to 11 strains per group. Four strains had unique clade assignment patterns. No obvious geographic pattern was observed in this analysis (Table 3 and Fig. 1).
Analysis in RDP4 confidently identified four genetic reassortment events. Reassortment was detected in strains Da1157, Da836, Da1166, and Da420. In all strains, except Da1166, seven methods (of the nine analyzed) detected reassortment (Supplementary Table S2). In the case of Da1166, six of the nine detected reassortment (all P values <0.02). All reassortment events were confirmed by the phylogenetic trees (Figs. 2–5). All parental strains implicated in each reassortment event were within 18 miles of the reassortant virus except for the event involving Da836. The suggested major parental strain, Da318, was collected nearly 50 miles from where Da836 was collected. However, the program was not confident in the identification of the parental strain and suggested it could be Da744, which was collected in the same location of Da836. The event detected in Da1166 suggests recombination instead of reassortment, as the breakpoint was detected in the middle of the segment 9 sequence, instead of at the end of segment 9 and beginning of segment 10 sequences.
Discussion
From 2008 to 2012, the Montana Health and Human Services Epidemiology section reported an average of one case of CTF per year in Montana (Baldry 2017). Three of the five reported cases were in Missoula County. Portions of Missoula County overlap with the BNF and areas where ticks were collected for this study. Most of the forest is within Ravalli County, which had no reported cases throughout the aforementioned time frame. We believe these numbers are a gross underrepresentation of the actual CTF cases within the state, most specifically the valley, and many cases of this disease are likely unreported or undiagnosed. The prevalence (6.7%) of D. andersoni infected with CTFV in the Bitterroot Valley would suggest there is a risk of infection by tick bite and possibly more human cases of CTF. For example, Rickettsia rickettsia has a prevalence of 1% in D. andersoni in the valley and has a higher number of reported cases within Montana, most likely due to the severity of the disease (Gage et al. 1994). In a recent report, the annual incidence of CTFV infection was determined for Ravalli and Missoula Counties at 9 and 5 per million, respectively (Yendell et al. 2015). In the same study, Sublette, Wyoming, had a noticeably higher incidence (141.9 per million) than all the other counties in five western states (Yendell et al. 2015). This county performed active surveillance for CTF, likely accounting for the higher incidence, again supporting that many cases likely go undiagnosed in areas where CTFV is endemic.
Segment reassortment has been widely documented in many human pathogenic RNA viruses, such as viruses belonging to the genus Orthonairovirus, Orthobunyavirus, Influenzavirus A, and other Reoviruses such as Rotavirus A and Bluetongue virus (McDonald et al. 2016). The influenza viruses and reoviruses package their virions with only one copy of each segment (McDonald et al. 2016). Reassortment occurs when a cell is coinfected with at least two strains of a segmented RNA virus and, during packaging, segments from each are incorporated into a new virion (Vijaykrishna et al. 2015). Influenza A, Rotavirus A, and Bluetongue virus use ribonucleoprotein (RNP) complexes and packaging signal sequences to select the required number of segments for packaging (McDonald et al. 2016, Burkhardt et al. 2014). Rotaviruses and Bluetongue virus are the best studied members of the Reoviridae family, in which CTFV resides. The inner core shell has 12 fivefold axes, which are believed to only contain space for 12 RNP complexed segments, exactly the number the CTFV contains (McDonald et al. 2016).
Reassortment has been only minimally documented for CTFV (Brown et al. 1989). Very few sequences exist, making analysis of CTFV reassortment difficult. As CTFV is a reovirus, a genus with many species capable of reassortment, it is probable CTFV can reassort. We found evidence of reassortment of CTFV strains found in ticks in the Bitterroot Valley. Reassortment occurred between strains from ticks collected from the southernmost and the northwestern region of the forest, a minimum distance of 30 miles away. Multiple strain types also appear to be circulating in close geographic proximity to each other, such as in the Blodgett and Mill Creek drainages, increasing the chance of coinfection and reassortment.
Since sequencing was performed on RNA extracts from single tick homogenates, some of the ticks may have been coinfected with multiple strains of CTFV and segments were sequenced from different virus strains in one tick. Although one would anticipate finding reassortants of CTFV, whether the data show reassortment or cocirculation of multiple CTFV strains within one tick, it emphasizes that the opportunity of reassortment for CTFV exists.
The mode of dispersal of strains is likely due to the host range overlap of small mammals moving infected ticks. Small mammals are the primary reservoir for CTFV and are readily fed on by wood tick nymphs and larva (both of which have a broad host range) and become persistently infected for life after an infected blood meal. It is not known if large mammals, such as deer and elk, produce a viremia and transmit the virus to ticks. If large mammals do become viremic, migration of infected animals could move CTFV genotypes around the valley. However, large mammals are not likely involved in long-distance dispersal of infected ticks in the valley because CTFV is not transovarially transmitted by D. andersoni and large animals are almost exclusively fed on by adult ticks (Eklund et al. 1959). Birds can play a role in the dispersal of infected ticks (Sparagano et al. 2015), however, birds are not the usual host for immature D. andersoni and the susceptibility of birds to CTFV infection is not known.
Conclusions
We conclude that the prevalence of CTFV infection in D. andersoni in the Bitterroot Valley and BNF is similar to that found during studies in the 1950s and 1960s, although prevalence varied by location sampled. We also found evidence of possible segment reassortment. Further studies should address the issue of reassortment by sequencing all segments or larger sections of those segments examined. The fragment lengths of segments 10 and 11 analyzed in this study were less than 500 bases. Therefore, the lack of variability between the strains could be due to the small regions examined. Recent improvements in next-generation sequencing will make sequencing larger regions more attainable. Sequencing should be performed on viral isolates obtained from ticks and small mammals, as opposed to total RNA extractions, to rule out sequencing mixed strain segments in coinfections. Attaining samples from a broader geographic range (sites across western states known to harbor D. andersoni and CTFV) would also assist in reassortment analysis of CTFV. The role of small rodents and large mammals in the infectious cycle of CTFV should also be studied, including the prevalence and strain composition in the animal population. A better understanding of CTFV within the tick and rodent population locally and regionally will better inform the public to the risk and improve mitigation practices.
Footnotes
Acknowledgments
This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health. We thank Ashley Kelly Palacio, Forest Hoyt, Sandra Stewart, and Paul Policastro for field help, Mike and Michael Zielinski and employees of the BNF, USDA, for collecting ticks, Austin Athman for help with figures, and Stephanie Seifert for article review.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.