
Abstract
In July 2016, a virus strain (JCC12-12) was isolated from Culicoides collected in Jiangcheng County, Yunnan Province, China. The JCC12-12 virus was sequenced using next-generation sequencing; a 9294-nt-long sequence with a G + C content of 35.63% was obtained by assembly and splicing. Phylogenetic analysis of the RNA-dependent RNA polymerase domain (RdRP) amino acid sequence showed that JCC12-12 virus shared the evolutionary branch of the Fort Crockett virus strain 22503a, isolated from Paracoccus marginatus in the United States; these viruses shared 87.5% homology, while the nucleotide homology of the JCC12-12 virus with other negeviruses was <50.8%, suggesting that JCC12-12 virus isolated from Culicoides was Fort Crockett virus. This is the first report of the Fort Crockett virus isolated from Culicoides.
Introduction
Negeviruses are single-strand, positive-strand RNA viruses. Their genomes are 9–10 kb in length and contain three coding frames: ORF1, ORF2, and ORF3. There are 17 known Negevirus species, which belong to two genera: Nelorpivirus and Sandewavirus. Multiple negeviruses have been isolated in North America, South America, Europe, Africa, the Middle East, and the Pacific islands; these viruses include Negev, Ngewotan, Piura, Loreto, Dezidougou, Santana, Tanay, and Okushiri viruses (Vasilakis et al. 2013, Kawakami et al. 2016, Fujita et al. 2017, O'Brien et al. 2017). In East Asia, several negeviruses have been isolated from mosquitoes collected in China, the Philippines, and Korea, as well as from Aedes larvae in Japan (Vasilakis et al. 2013, Nabeshima et al. 2014, Hang et al. 2016, Kawakami et al. 2016, Fujita et al. 2017, Wang et al. 2018). These results indicate that the geographic distribution of negeviruses is considerably wide and that negeviruses occur in mosquitoes from tropical and temperate regions.
Negevirus is a recently proposed new taxon of insect-specific viruses with a wide host range in Diptera (Vasilakis et al. 2013, Carapeta et al. 2015, Nunes et al. 2017). Many negeviruses have been isolated from field mosquitoes and phlebotomine sand flies in the genus Lutzomyia. Within the family Culicidae, nine genera of mosquitoes (Aedes, Culex, Anopheles, Armigeres, Psorophora, Uranotaenia, Deinocerites, Wyeomyia, and Trichoprosopon) have been reported to exhibit natural infection with negeviruses; the natural infection rates and infection titers of these viruses are substantial in some mosquito species (Vasilakis et al. 2013, Carapeta et al. 2015). Molecular analysis has demonstrated that most negeviruses exhibit high genetic variability, wide host ranges, and cross-species transmission in a variety of insect vectors (Nunes et al. 2017). In 2015, a new Negevirus, Fort Crockett virus, was isolated from Paracoccus marginatus (mealy bug) collected in the United States (Nunes et al. 2017), suggesting that other types of insects may also be naturally infected with negeviruses. In this study, a strain of Fort Crockett virus was isolated from Culicoides collected in Yunnan Province, China, in 2016.
Culicoides and mosquitoes were collected from cattle shelters using light traps (12 V, 300 mA; Wuhan Lucky Star Environmental Protection, Hubei, China) in a village in Jiangcheng County, Yunnan Province, on the Chinese side of the China–Laos–Vietnam border in July 2016. Culicoides and mosquitoes were collected overnight, from 7:00 pm to 7:00 am. Based on morphological characteristics, female Culicoides were selected from the captured insects. A total of 6600 Culicoides divided into 66 pools were collected, but no mosquitoes were collected, from a cattle farm in a village of Jiangcheng County. The Culicoides pools were homogenized in 1 mL of grinding fluid (minimum Eagle's medium [MEM] [pH 7.4] containing 100 U/mL of penicillin and 100 μg/mL of streptomycin) in a sterile glass grinder and centrifuged; the supernatant was then inoculated on monolayers of C6/36 cells in 24-well plates and blind-passaged three times (7 days per cycle), as previously described (Wang et al. 2015, 2017). The JCC12-12 strains caused cytopathological effects (CPE) in the third blind passage in C6/36 cells, 120 h after inoculation. The CPE were characterized by cell refractive enhancement and clustering. No CPE were observed in mammalian BHK-21 or Vero cells.
Genomic DNA of the insect was extracted from the grinding suspension of JCC12-12 pool, and the COI gene was amplified by PCR (Dallas et al. 2003, Yuan et al. 2019) and sequenced. Sequence analysis showed that this pool of midges was mainly Culicoides sumatrae.
The viral RNA was extracted from C6/36 cells infected with an isolate (JCC12-12) using RNAiso Plus (TaKaRa, Dalian, China), in accordance with the manufacturer's instructions. First-strand cDNA was synthesized using the specific primer K2Sr (5′-GACCATCTAGCGACCTCCACNNNNNN-3′) (20 μmol/L) (synthesized by Sangon, Shanghai, China) and PrimeScript® Reverse Transcriptase (TaKaRa), as previously described (Zhang et al. 2015, Wang et al. 2018). The complementary cDNA was synthesized using 2.5 U of Klenow enzyme (TaKaRa), after initial denaturation at 94°C for 2 min, followed by incubation at 37°C for 1 h and inactivation at 70°C for 10 min. Random PCR was performed with primer K2S (5′-GACCATCTAGCGACCTCCAC-3′) (20 μmol/L) (synthesized by Sangon). The amplified products were separated by 1% agarose gel electrophoresis with 1 × TAE buffer (40 mM Tris-acetate, 1 mM EDTA, pH 8.0) with GoldView nucleic acid dye. PCR fragments >500 bp were purified using the TaKaRa DNA Fragment Purification Kit ver. 2.0 (TaKaRa) and sequenced by next-generation sequencing. The raw next-generation sequencing reads were subjected to quality control and trimming procedures using CLC Genomics Workbench 11 software, followed by de novo assembly and identification of viral contigs using blastx. A 9294-nt-long sequence of the JCC12-12 virus was obtained with a G + C content of 35.63%.
Nucleotide homology analysis showed that the JCC12-12 strain had the highest homology (87.5%) with Fort Crockett virus strain 22503a, isolated from P. marginatus in the United States in 2015. Nucleotide homologies of the JCC12-12 strain with other negeviruses (e.g., Tanay, Wallerfield, Loreto, and Piura viruses) were 35.9–50.8%, whereas they were 36.739–43.9% with plant viruses, Hibiscus virus and Ligustrum Citrus virus. Open reading frame (ORF) analysis predicted three ORFs within the JCC12-12 virus, ORF1, ORF2, and ORF3, which were 6948, 1002, and 420 nt in length, encoding 2215aa, 333aa, and 139aa, respectively. Similar to negeviruses (Nabeshima et al. 2014, Wang et al. 2018), four conserved domains were identified in ORF1: a viral methyltransferase domain (vMet), an FtsJ-like methyltransferase (ribosomal RNA methyltransferase) domain (FtsJ), a viral helicase domain (Hel), and an RNA-dependent RNA polymerase domain (RdRp) (Fig. 1). The ORF1 and ORF2 (partial sequence, 837 nt) nucleotide homologies of the JCC12-12 virus with Fort Crockett virus strain 22503a were 91.5% and 92.9%, respectively; the respective amino acid homologies were 97.2% and 96.1%. In contrast, the nucleotide homologies with other negeviruses were lower than 50.6% and 45.6%, respectively; the respective amino acid homologies were lower than 48% and 20%. The ORF3 nucleotide and amino acid homologies of JCC12-12 virus with other negeviruses were less than 37% and 30%, respectively; no Fort Crockett virus strain 22503a ORF3 sequence is present in GenBank. Nucleotide sequence data reported here are available in the GenBank database under accession numbers MN901928. Phylogenetic analysis based on the RdRP amino acid sequence demonstrated that the JCC12-12 virus isolated from Culicoides sumatrae was Fort Crockett virus (Fig. 2). To the best of our knowledge, this is the first report of the Fort Crockett virus isolated from Culicoides in China.
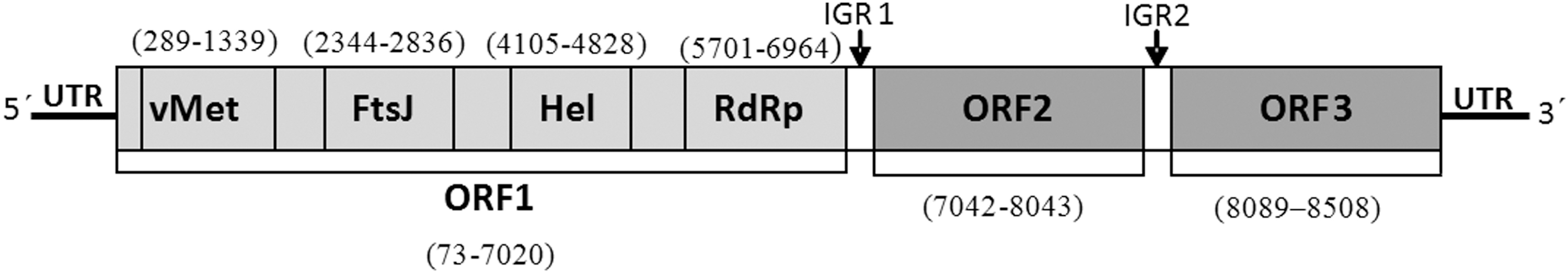
The genome organization of Fort Crockett virus isolated from China. vMet, a viral methyltransferase domain; FtsJ, an FtsJ-like methyltransferase (ribosomal RNA methyltransferase) domain; Hel, a viral helicase domain; RdRp, an RNA-dependent RNA polymerase domain; UTR, untranslated region; IGR, intergenic region.

ML phylogenetic trees of RdRP region (1877–2298), amino acid sequences of Fort Crockett virus isolated from China. The best substitution model is LG+G.
A pair of primers (CRV884F: 5′-ACGAGGGGTTATGGCTACG-3′; CRV1898R: 5′-CACTAAGCCCTAATGCGACC-3′) was designed based on the sequence of JCC12-12 strain, and 66 pools of Culicoides and 19 isolates from them were detected by RT-PCR. The results show that JCC12-12 was positive, and the other 65 pools were negative. At the same time, 18 isolates from Culicoides collected at the same place and at the same time point with JCC12-12 were also subjected to sequence by next-generation sequencing. There were only about 700 and 800 reads matching with the Fort Crockett virus in two isolates, respectively, but RT-PCR detection for the two isolates was negative, indicating that the two isolates from Culicoides may also exist as Fort Crockett virus, but the virus titer is very low, which is difficult to detect by RT-PCR.
Many species of mosquitoes, phlebotomine sand flies, and P. marginatus are naturally infected with negeviruses (Vasilakis et al. 2013, Nunes et al. 2017). Given their worldwide distributions and broad host ranges in these insects, negeviruses likely infect other types of insects. In this study, a variant strain of Fort Crockett virus was isolated from Culicoides collected in Yunnan, China, indicating that Culicoides may be naturally infected with Fort Crockett virus or (potentially) with multiple negeviruses. This finding is important for efforts to understand the geographic distribution, genetic diversity, and evolution of negeviruses. Furthermore, negeviruses are insect-specific viruses with mosquitocidal activity (Carapeta et al. 2015); Fort Crockett virus may therefore be an attractive candidate for use in biological control of Culicoides, with the aim of reducing the transmission of Culicoides-borne diseases.
Phylogenetic analysis demonstrated that the Fort Crockett virus isolated from P. marginatus collected from the United States was genetically distinct from the nelorpiviruses and sandewaviruses, but phylogenetically closer to plant viruses in the genera Higrevirus, Blunevirus, and Cilevirus (Kallies et al. 2014, Nunes et al. 2017). Similarly, the Fort Crockett virus isolated from Culicoides in this study was genetically and evolutionarily related to these plant viruses. Whether this virus is pathogenic to plants and whether the transmission of other vectors influences the transmission of plant or animal pathogens should be assessed in future studies.
Negevirus has only been discovered and recognized in recent years, and it is widely distributed in the world. In recent years, two kinds of negeviruses have also been found in China. Several strains of Tanay virus were isolated from Culex tritaeniorhynchus, C. quinquefasciatus collected in Guangxi in 2013 (Wang et al. 2018), and Anopheles sinensis in Yunnan in 2015 (Zhao et al. 2019), and a Manglie virus from Culex tritaeniorhynchus in Yunnan in 2011 (Wang et al. 2019). These negeviruses have been isolated from mosquito samples in the last several years. In this study, no mosquito samples were collected in a cattle farm in a village of Jiangcheng County, Yunnan province, so the Fort Crockett virus was not detected in mosquito. The collection site of Culicoides has good vegetation and high humidity, and it is suitable for the growth and reproduction of Culicoides. The density of Culicoides is very high in the cattle farm, and Culicoides are the main blood sucking insects in cattle pens in this area. Fort Crockett virus has been isolated or detected from Culicoides collected in the cattle farm of Jiangcheng County. These results indicated that negevirus was widely detected in mosquitoes, midges, and other vector insects in southern China. We should strengthen the study on the influence of negevirus on the transmission of human or animal pathogenic arboviruses.
Footnotes
Acknowledgment
We thank Professor Wu Aiping at the Suzhou Institute of Systems Medicine for data analysis.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work is supported by grants from the National Major Research and Development Projects (grant no. 2016YFD0500300), the National Natural Science Foundation of China (grant no. 31660714 and 31460669), Basic Research Projects of Yunnan Province (grant no. 2019FA015), Yunnan Science and Technology Talents and Platform Plan (grant no. 2018HB046), and the Yunnan Animal Science and Veterinary Institute Applied Basic Research Projects (no. 2019RW006, 2019RW007, 2019RW009). The funders had no role in the study design, data collection and analysis, and in the decision to publish or preparation of the article.