
Abstract
Borrelia burgdorferi sensu lato (s.l.) is the most common pathogen of medical significance transmitted by ticks of the family Ixodidae in Belarus. Human infection with B. burgdorferi causes Lyme borreliosis, most commonly referred to as Lyme disease. Currently, 20 species of Lyme disease-associated Borrelia and more than 20 relapsing fever-associated Borrelia species have been identified. These etiologic agents belong to the genus Borrelia in the family Spirochaetaceae. Genetically characterized isolates with specific sequences have proven that these pathogens are endemically transmitted in many European and Asian countries. In addition, joinpoint regression analysis is often applied to characterize infection trends over time and to identify the time point(s) at which the trend significantly changes. In this epidemiological investigation, joinpoint analysis was applied to investigate the temporal trend of B. burgdorferi s.l. infections in 4070 ticks collected between April and October 2012–2019. Detection of Borrelia species in ticks is an important tool to determine temporal and geographic distribution and abundance, and to predict the risk of Lyme disease to people in different regions. Our data provide a basis for further studies to determine the distribution and abundance of B. burgdorferi s.l. species in Belarus.
Introduction
Borrelia burgdorferi sensu lato (s.l.) is a group of spirochetes belonging to the genus Borrelia in the family of Spirochaetaceae. With respect to medical significance, as originally described, species in the Borrelia genus may be categorized in two groups based on the diseases that they can cause. One group includes organisms that are associated with relapsing fever, while the other group is commonly called the Lyme complex (formerly B. burgdorferi sensu lato, or s.l.). In recognition of group-specific genomic distinctions, a proposal has suggested that the genus should be divided and with Lyme disease pathogens being classified with a new designation, “Borreliella,” however, has not been universally endorsed (Adeolu and Gupta 2014). The designation is considered synonymous with the Lyme complex, Lyme Borreliosis group, Borrelia, B. burgdorferi s.l., Bb, and Borreliella and indicative of the Lyme disease (borreliosis) causing spirochetes. This debilitating illness historically is predominantly associated with the North American pathogen, B. burgdorferi sensu stricto (s.s). In Europe and Asia, it is now well-recognized that the widely distributed and emerging species Borrelia afzelii and Borrelia garinii are of increasing clinical relevance (Margos et al. 2018, Bamm et al. 2019). Both Reye et al. (2013) and Kniazeva et al. (2021) have described the wide distribution of Ixodes ricinus and Dermacentor reticulatus in Belarus and the presence of several tick-borne pathogens, including Anaplasma phagocytophillum, Babesia microti, Babesia venatorum, Bartonella henselae, B. afzelii, B. burgdorferi s.l., Borrelia miyamotoi, Coxiella burnetii, Ehrlichia muris, Francisella tularensis, and tick-borne encephalitis virus.
Hard ticks (Acari: Ixodidae) that are the principal vectors of viral, bacterial, and protozoan zoonotic diseases are abundant and widely distributed in the Northern hemisphere and can be infected by numerous different microorganisms (European Food Safety Authority 2010, ECDC 2019) and effectively transmit some of these simultaneously to vertebrates causing coinfection (Diuk-Wasser et al. 2016). Tick-borne diseases are recognized as a significant and increasing public and animal health issue worldwide. Data on these diseases and tick vectors in Belarus are very limited. Belarus was included in the 2017 review of vector-borne diseases risks to the European Union (Braks et al. 2017). The transmission cycle of B. burgdorferi s.l. is complex since it depends on the interactions of the vectors with the reservoir hosts and the pathogenic agents, all of which are influenced by several biotic and abiotic factors that vary in space and time (Wint et al. 2018). Understanding the cycle and collection of data on the pathogen and vectors is critical for disease diagnosis and for risk management. The primary tick vector associated with Lyme disease outside of North America, namely I. ricinus, is widely distributed (Seasonal active surveillance for ticks over European Centre for Disease Prevention and Control Tick-borne encephalitis, 2019—
The date presented here demonstrate the utility of polymerase chain reaction (PCR) as a molecular screening tool for the identification of infected ticks and genotyping of Borrelia. Before the development of molecular protocols, infections in ticks in Belarus were determined by analysis of the tick gut contents using anti-Borrelia immunofluorescence (IF) staining and microbial culture (Vedenkov et al. 2008). Inherent limitations of these approaches include the relatively slow growth and fastidious requirements of B. burgdorferi s.l., performance issues associated with antibodies, and the level of expertise required for IF processing. Consequently, PCR was utilized as the best approach to provide rapid, sensitive, and specific identification of infected vectors. This molecular technique offers notable advantages over traditional methodologies. These include direct culture-independent application, genotyping, and simultaneous detection of multiple pathogens in ticks. In this study, we used the combination of qPCR and nested PCR (nPCR) for the Borrelia spp. with primers to Flagellin B (FlaB) (Reye et al. 2013).
Specific and strategic gene selection and the design of amplicons are critically important to ensure experimental success. The primers designed for the purpose of accurately estimating the presence of B. burgdorferi s.l., must therefore detect the relevant pathogens without cross-reacting with relapsing fever spirochetes also in the Borrelia genus. The FlaB gene that encodes a major protein of the flagellum is located on the single linear chromosome. With respect to FlaB, this specificity is achieved by targeting a variable internal region of FlaB, as opposed to targeting more conserved flanking sequences that are commonly shared by diverse bacteria. The 5′ and 3′ termini of the FlaB gene are shared by organisms beyond B. burgdorferi s.l. As a result, these regions are unsuitable for the specific assessment of the pathogens that cause Lyme disease. As described by Wills et al., (2018), when using the less-conserved interior sequences as primer targets, two rounds of PCR are required to detect a final, internal amplicon.
The aim of the present study was to provide insight into the diversity and prevalence of B. burgdorferi s.l. in Belarus carried/transmitted by Ixodid ticks and to analyze the dynamic of the incidence of the pathogen over time. The studies described here reveal the widespread distribution of B. burgdorferi s.l. in ticks and present new data on its prevalence.
Methods
Belarus is a landlocked country of eastern Europe with a population of 9.4 million, 78.4% of which reside in urban areas bordered by Lithuania and Latvia to the north west, by Russia to the north and east, by Ukraine to the south, and by Poland to the west. The country of Belarus is divided into six administrative districts (Brest, Gomel, Grodno, Minsk, Mogilev, and Vitebsk regions), each centered around a major city (Minsk). Much of the country consists of flat lowlands separated by low-level topped hills and uplands. The terrain is relatively flat; the highest point, Dzyarzhynskaya Hill, being only 1135 feet (346 meters) above sea level. Over half of the surface area of Belarus lies below 660 feet (200 meters), and about 40% of the country is forested. The most common tick species in Belarus are I. ricinus and D. reticulatus (Reye et al. 2013, Kniazeva et al. 2021).
Tick collection
Ticks were collected by dragging a 1 m2 white canvas cloth along the ground in wooded and grassy habitat (Vassallo et al. 2000) during the season of peak activity between April and October 2012–2019 in all administrative regions of the territory of the Republic of Belarus (Fig. 1). Adult ticks were identified to species, and then pools of up to 10 individual ticks were maintained alive in collection tubes, with moist filter paper and sand to provide a suitably humid environment to keep the ticks alive until processing. Tubes were labeled with species, location, and date of collection.
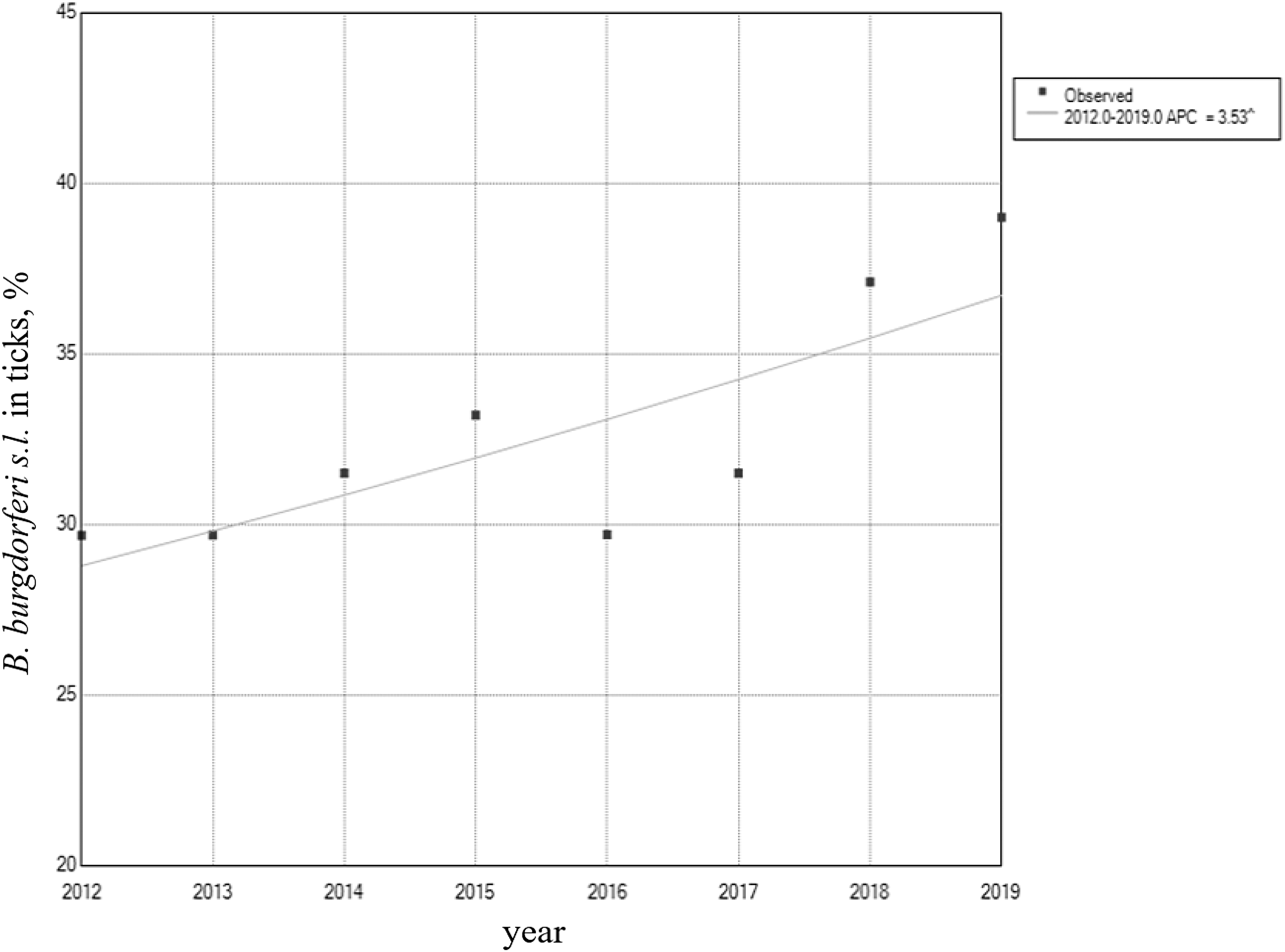
Joinpoint regression model of Borrelia burgdorferi s.l in ticks in Belarus. ^indicates that the Annual Percentage Change (APC) is significantly different from zero at the alpha = 0.05 level. Final Selected model: 0 Joinpoints.
Tick processing and DNA extraction
After delivery to the laboratory, ticks were washed in 70% ethanol and phosphate-buffered saline (PBS) buffer (pH 8.0), sorted individually into vials labeled with information on species, sex, the collection site, and date of collection. The ticks were then homogenized by grinding using a XENOX motor handpiece MHX/E (PROXXON, Gmb).
Nucleic acids were extracted from individual ticks using the AmpliSens® RiboPrep reagents kits (InterLabService Ltd., Russian Federation) following the manufacturer's instructions. cDNA was obtained by using Reverta-L® kit (InterLabService Ltd., Russian Federation) following the manufacturer's instructions. All ticks were analyzed by real-time PCR amplification for the presence of B. burgdorferi s.l., using the commercial kit BelarTBD-PCR/RT® (RRPCEM, Belarus) in Bio-Rad CFX96 machine (Semizhon et al. 2014).
RT-PCR-positive tick extracts were further tested with a nPCR assay designed to amplify a portion of the highly conserved 41-kDa chromosomal flagellin (flaB) gene of Borrelia species. Reaction mixtures for single-stage PCRs and first-round amplifications of nPCR assays contained 2.5 μL buffer, 1 μL MgCl2, 0.5 μL dNTP, 10 uM Outer 1 primer (5′-AARAATTGGCAGTTCAATC-3′), 10 μM Outer 2 primer (5′-GCATTTTCWATTTTAGCAAGAAGTGATG-3′)—for the first round, and 10 μM Inner 1 primer (5′-ACATATTCAGATGCAGACAGAGGTTCTA-3′), 10 μM Inner 2 primer (5′-GAAGGTGCTGTAGCAGGTGCTGGCTGT-3′) for the second round, 2.5 U Taq polymerase, and 5 μL of DNA extract per individual sample in a total reaction volume of 25 μL. The thermocycling conditions consisted of initial denaturation at 95°C for 1 min, followed by 40 cycles of 94°C for 30 s, primer annealing at the temperature 68°C for 30 s (for the first round) and 59°C for 30 s (for the second round), and extension at 72°C for 30 s. Mixtures for nested reactions included between 1 and 2.5 μL of outer reaction product as the template. Amplicons were visualized on 2% agarose gels stained with ethidium bromide and were documented with a digital gel imaging system (Gel Doc XR+ Gel Documentation System, Bio-Rad). Size of the amplicons was 497 bp for the first round of nPCR and 389 bp for the second round (Clark et al. 2005.).
DNA sequence analysis
PCR products were purified using NucleoSpin Gel and PCR Clean-up, Mini kit for gel extraction, and PCR clean up purification kit (MACHEREY-NAGEL, DE). DNA templates were sequenced using the fluorescent dideoxy terminator method in genetic analyzer ABI PRISM® 3100-Avant manufactured by Applied Biosystems using the company's reagents. Sequences were generated using Sequencher Software (Gene Codes Corporation, Ann Arbor, MI). Investigator-derived sequences were compared with those obtained by searching GenBank database (National Center for Biotechnology Information) using Basic Local Alignment Search Tool (BLAST) and aligned using Clustal X. Phylogenetic trees were constructed using neighbor-joining (NJ) and maximum parsimony (MP) methods with the tree-building program MEGA version 2.1. Tree topologies and genetic relationships obtained with the two methods were compared for consensus. To estimate node reliability of trees obtained with each method, bootstrap values based on an analysis of either 100 (MP) or 1000 (NJ) replicates were determined. Pairwise distances were computed by the Kimura two-parameter model.
Statistical analysis
To determine whether or not changes in the B. burgdorferi infection rates of ticks followed a trend over time, joinpoint regression was estimated for annual percentage of infected ticks group by using the Joinpoint Trend Analysis Software, Version 4.5.0.1 (Statistical Research and Applications Branch, National Cancer Institute;
In brief, by using annual percentage of infected ticks rate data as inputs, this method identifies the year(s) when a trend change occurs. One can therefore calculate the annual percentage change (APC) in rates between trend-change points, and also estimate the average annual percentage change (AAPC) in the whole period studied.
To estimate the APC, the following model was used:
log (Yx ) = b 0 + b 1 x, where log(Yx ) is the natural logarithm of the rate in year x.
Then, the APC from year x to year x + 1 is:
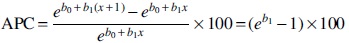
When there are no join points (i.e., no changes in trend), APC is constant, so it equals the AAPC (Radzišauskienė et al. 2018, Dragomirescu et al. 2019).
Results
Sampling was conducted in every administrative unit of the Republic of Belarus. A total of 4070 questing adult Ixodid ticks were collected, comprised 3320 (82%) I. ricinus and 750 (18%) D. reticulatus (635—year 2012; 576—year 2013; 469—year 2014; 750—year 2015; 552—year 2016; 504—year 2017; 311—year 2018, 272—year 2019). Ticks were individually PCR-analyzed for the presence of DNA of B. burgdorferi s.l. The average yearly infection rate for the study period was 32.4% with the lowest rate (29.67%) in 2012 and the highest (39%) in 2019. Table 1 shows the observed and predicted infection rates for ticks and the percent changes, respectively.
Prevalence of Borrelia burgdorferi s.l in Ixodes ricinus and Dermacentor reticulatus in Belarus in Year 2012–2019
B. burgdorferi s.l., Borrelia burgdorferi sensu lato.
General trend in B. burgdorferi s.l. prevalence in ticks
To analyze trends of B. burgdorferi s.l. infected ticks over time and to determine if observed trend patterns were significant, we applied joinpoint regression analysis. Figure 1 displays the increasing trend of B. burgdorferi s.l. infected ticks by 3.5% every year, and that was significantly different from zero at a = 0.05 (Fig. 1 and Table 2).
Annual Percentage Change
Indicates that Annual Percentage Change (APC) at significantly different from zero at a = 0.05.
APC, annual percentage change; CI, confident interval.
Genospecies diversity of B. burgdorferi s.l. in ticks in Belarus
The study was conducted on 40 B. burgdorferi s.l. randomly selected positive I. ricinus ticks, collected in Minsk region—the most populated area of Belarus. We sequenced nPCR-amplified flaB gene fragments from tick samples and compared the sequences to those obtained via BLAST searching the GenBank database. Most of the flaB DNAs analyzed in this study were sequenced in one direction only, with the forward primers used in the PCRs. Based on the BLAST scores and phylogenetic comparisons we conducted, all flaB DNAs obtained from ticks belonged to B. burgdorferi s.l. The analyses revealed that the 18 strains (40%) typed in this study were definitely B. afzelii strains. The next most common genospecies detected was B. burgdorferi s.s. In comparison, the species B. garinii and Borrelia valasiana were present in relatively small numbers (Table 3). Fig. 2shows a NJ phylogenetic tree based on 389 bp of the flaB gene sequences comparison, which has been used to differentiate the genospecies (1) B. afzelii, (2) B. burgdorferi s.s., (3) B. garinii, and (4) B. valasiana at intraspecies level according to their origins.

The genetic analysis with NJ method on Borrelia burgdorferi s.l isolates showed clustering according to reference sequences.
Species of Borrelia burgdorferi s.l Isolated from Ixodes ricinus Ticks from Minsk Region
B. burgdorferi s.s., Borrelia burgdorferi sensu strict.
The present B. afzelii isolates of Belarus are closer to Russia samples than the ones from the U.S., China, Poland, Switzerland, Japan, and Turkey. Interestingly, B. afzelii from Russia formed a separate clade on the phylogenetic tree and genospecies from Belarus and other references formed another clade (Fig. 2a).
The B. burgdorferi s.s. references from Belarus constitute a cluster between the first group (group1) involving France, Serbia, China, U.S., Russia, Turkey, Poland, and Canada sequences and another clade with the strains from Russia. The MSQ550 B. burgdorferi s.s. Belarus has the closest similarity to Russia isolates. MSQ 553 B. burgdorferi s.s. Belarus is genetically close to the references from China, France, and Poland (Fig. 2b). MSQ1023 and MSQ24 B. burgdorferi s.s. Belarus lined up with the spirochetes reported in Canada and Poland. This cluster is genetically different from the one involving Serbia, U.S., Russia, Turkey, Poland, and China strains.
The B. garinii Belarus isolate constitute a single clade close to references from Poland and genetically different from other all reference genospecies with long distance (Fig. 2c).
The B. valasiana we analyzed is very close to Poland strains too but located in a different branch distant from other reference strains comprising East Asian countries in the phylogenetic tree (Fig. 2d).
Discussion
The hard tick I. ricinus serves as the main vector for spirochetes of the B. burgdorferi s.l. complex in Eastern Europe (Eisen et al. 2020). In this study, the circulation of several B. burgdorferi s.l. species was confirmed over a wide geographic range, covering most of Belarus. According to our investigation, on average 32.4% of ticks were infected with B. burgdorferi s.l. in the years 2012–2019. The Annual Percent Change (APC) for the pathogen significantly increased by 3.5% every year. Sequencing analysis revealed four pathogenic genotypes of the genus Borrelia with the prevalence of B. afzelii at 40%. This demonstrates the endemic presence of Borrelia spp. in ticks in Belarus. Phylogenetic analysis revealed local subpopulation of B. afzelii isolates and mixed population of B. burgdorferi s.s. isolates that could be explained by bird migration routes passing through territories of the country (Jourdain et al. 2007). Out of 323 species of birds ever recorded in our country, only 39 live here settled, 232 species migrate in spring and autumn at different times and in different numbers, and the rest—91 species—fly to Belarus by accident or arrive for the winter (Nikiforov et al. 2013).
It is important to take into account the possible increased exposure of people to tick bites in the Republic of Belarus, and to provide appropriate information to the institutions, physicians, forestry workers, and visitors. They can then adopt proper precautions when ticks are active (Pintore et al. 2015) and assist with the implementation of effective control strategies for the management of ticks and pathogens that they transmit.
Footnotes
Acknowledgments
The authors acknowledge entomology departments of the Republican and Regional Centers for Hygiene, Epidemiology and Public Health of the Republic of Belarus for the assistance in ticks collection, Lyme disease study group (ESCBOR), and The European Society of Clinical Microbiology and Infection Diseases for their encouraging research and financial support in attending ESCMID summer schools and ECCMIDs to present and discuss the results, and the United States Department of Defense, Defense Threat Reduction Agency (DTRA), Cooperative Biological Engagement Program (CBEP) for their assistance and financial support in publication of this article. While DTRA/CBEP did not support the research described in this publication, the Program supported the article publication. The contents of this publication are the responsibility of the author and do not necessarily reflect the views of DTRA or the United States Government.
Author Disclosure Statement
No conflicting financial interests exist.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.