
Abstract
Background:
In August 2013, a virus strain (DH13M98) was isolated from Culex tritaeniorhynchus Giles collected in Mangshi, the southwestern border area of Yunnan Province, China. The virus replicated and caused cytopathic effects (CPE) in Aedes albopictus (C6/36) cells, but not in baby hamster Syrian kidney (BHK-21) cells.
Materials and Methods:
Agarose gel electrophoresis (AGE) analysis revealed that the DH13M98 virus was a 10-segment double-stranded RNA (dsRNA) virus, with a “1-1-1-2-1-1-2-1” pattern. The full genome of the DH13M98 virus was sequenced by full-length amplification of complementary DNAs (FLAC).
Results:
Phylogenetic analysis of the viral RNA-dependent RNA polymerase (Pol), major subcore-shell (T2), and major core-surface (T13) protein showed that DH13M98 clustered with Umatilla virus (UMAV), and the amino acid (aa) sequences of DH13M98 shared more than 89.5% (Pol), 95% (T2), and 91.1% (T13) identity with UMAV. However, the aa identity of outer capsid protein one (OC1) of DH13M98 with other UMAV was 57.1–79.2%, suggesting that DH13M98 was UMAV, but distinct from other strains of UMAV from the United States, Japan, and Germany at OC1, and it may be a high variant strain of UMAV, even a new serotype.
Conclusion:
This is the first isolation of UMAV in China, which enriches the resources of virus species in China and provides new insights into the genetic diversity and geographical distribution of the virus.
Introduction
The orbivirus is a genus of viruses in the family Reoviridae, which currently includes 22 species (representing 22 distinct virus serogroups) that have been recognized by the International Committee for the Taxonomy of Viruses (ICTV), as well as several closely related viruses that have not been approved as species but are being considered potential members (Mertens et al. 2005, Vieira et al. 2009, Attoui et al. 2011). Orbiviruses are widely distributed in the world and have been isolated from human and various animals, such as domestic and wild ruminants, equines, marsupials, sloths, bats and birds, and from hematophagous arthropods, including ticks, midges, and mosquitoes (Karabatsos 1985, Attoui et al. 2009, Belaganahalli et al. 2011a).
Several orbiviruses, such as bluetongue virus (BTV), African horse sickness virus (AHSV), and epizootic hemorrhagic disease virus (EHDV), are important pathogens of animals and cause severe hemorrhagic disease in sheep, cattle, horse, deer, or other ungulates, which have a serious impact on the livestock industry (Attoui and Mohd Jaafar, 2015, Dennis et al. 2019, Maclachlan et al. 2019). Therefore, the three diseases are also listed as Class I by the World Organization for Animal Health, and much attention has been paid to controlling and preventing of them (Murota et al. 2020).
Orbiviruses are nonenveloped viruses and have icosahedral symmetry (Attoui et al. 2011). The viral genome consists of ten double-stranded RNA (dsRNA) segments, designated Seg-1 to Seg-10 from largest to smallest, which encode seven distinct structural proteins (VP1–VP7) and at least three or four distinct nonstructural proteins (NS1–NS2, NS3/NS3a, and NS4) (Mohd Jaafar et al. 2014, Belaganahalli et al. 2015). Phylogenetic analysis based on Orbivirus protein sequence has demonstrated that orbiviruses are divided into three major clusters, mosquito-borne (MBOs), Culicoides/sand fly-borne (CBOs/SBOs), and tick-borne (TBOs), suggesting coevolution of orbiviruses with their arthropod vectors (Belaganahalli et al. 2014, Fagre et al. 2019).
Umatilla virus (UMAV) was first isolated from Culex pipiens collected in 1969 in Oregon, and then reisolated from Culex spp. and sparrows (Passer domesticus) collected in Colorado, Texas, and Utah in the United States during 1967–1970 (Belaganahalli et al. 2011b). Three other serotypes of UMAV, Llano Seco virus (LLSV), Minnal (MINV), and NETV (NETV), were subsequently isolated from mosquitoes collected in the United States, India, and Israel (Mertens et al. 2005, Attoui et al. 2011). Stretch Lagoon orbivirus (SLOV) was isolated from Culex annulirostris collected in Kimberley, Austria, in 2002 (Cowled et al. 2009), and Koyama Hill virus (KHV) was isolated from Culex sasai collected in Japan, in 2011 (Ejiri et al. 2014). Sequence analysis showed that both of them were UMAV (Belaganahalli et al. 2011b, Ejiri et al. 2014).
Recently, several strains of UMAV have also been isolated in Germany, suggesting that UMAV has a wide geographical distribution. In this study, a new UMAV (DH13M98) was isolated from Culex tritaeniorhynchus Giles collected in Mangshi, the southwestern border of Yunnan province, China, and its molecular properties and phylogenetic relationships with members of orbivirus were described in detail. This is the first time UMAV was isolated in China.
Materials and Methods
Mosquito collection
Mosquitoes were collected from bovine shelters at night using one light trap (220 V, 50 Hz, 8 W; Zhongshan Jili Electric Appliance Manufacturing Co., Ltd., Guangdong, China) in a suburb of Mangbing Village (24°20′2″ N, 98°32′13″ E, altitude 1101 meters) in Mangshi City, Dehong Prefecture, southwestern Yunnan Province on July 6, 2013 (Fig. 1). Captured mosquitoes were frozen to death in a −20°C refrigerator, then classified and identified on a chilled plate by using morphologic characteristics under a stereoscopic microscope. Every 50 mosquitoes of the same species were pooled in a cryogenic vial and stored in liquid nitrogen until virus isolation.

Local map of the mosquito sample collection sites in Mangshi City, Dehong Prefecture, southwestern Yunnan Province, China.
Cell cultures
Baby hamster Syrian kidney (BHK-21) and Aedes albopictus (C6/36) cells were used for virus isolation (Wang et al. 2015). BHK-21 were grown in minimal essential medium (MEM; HyClone) with a balanced salt solution supplemented with 10% fetal bovine serum (FBS), 100 U/mL of penicillin, and 100 μg/mL of streptomycin and were maintained at 37°C in 5% CO2. C6/36 were cultured in 45% Dulbecco's modified Eagle's medium (DMEM; HyClone) containing 10% FBS, 30% Roswell Park Memorial Institute (RPMI) 1640, 100 U/mL penicillin, and 100 μg/mL streptomycin, and were maintained at 28°C in 5% CO2.
Virus isolation
Virus isolation was performed according to a previously described method (Wang et al. 2015). Mosquito pools were homogenized in 1 mL of ice-cold MEM containing 100 U/mL of penicillin and 100 μg/mL of streptomycin, using a TissueLyser II (QIAGEN GmbH, Hilden, Germany). The homogenates were centrifuged at 5000 g for 10 min, and then 200 μL of supernatant was inoculated onto monolayers of BHK-21 and C6/36 cells in 24-well culture plates for 7 days. After three additional blind passages, the cells were observed daily for cytopathic effects (CPE) and the cell supernatants were harvested and kept at −80°C until analysis.
Viral dsRNA extraction and electropherotype analysis
Viral RNA was extracted from infectious C6/36 cells using RNAiso Plus (TaKaRa, Dalian, China) according to the manufacturer's instructions. Separation of viral dsRNA from the total RNA procedure was described by Attoui et al. (2000). Six microliters of viral RNA was taken for electropherotype analysis, and the remaining RNA was either used immediately or stored at −80°C. Viral genomic segments were separated by 1% agarose gel electrophoresis (AGE) at 80 V for 4 h in 1 × TAE buffer, stained with GoldView II (Solarbio, Beijing, China), and visualized under ultraviolet light.
Reverse transcription of dsRNA and PCR amplification of complementary DNAs
The genome segments of the virus were reverse-transcribed using a “full-length amplification of complementary DNA” (FLAC) technique described by Maan et al. (2007). The viral dsRNA genome segments were separated by 1% agarose gel in 1 × TAE buffer, and then, the dsRNA bands were excised from the gel in eight groups (Seg-1; Seg-2; Seg-3; Seg-4 and 5; Seg-6; Seg-7; Seg-8 and 9; Seg-10). Each purified viral dsRNA was ligated to anchor-primer using the T4 RNA ligase (TaKaRa), followed by reverse transcription using RT system. The resulting complementary DNAs (cDNAs) were amplified using complementary primers to the anchor-primer and for cloning purposes with a high-fidelity GXL DNA Polymerase (TaKaRa).
Cloning and sequencing of cDNAs
The amplicons of cDNA were analyzed by 1% AGE (100 V for 2 h). Each cDNA segment of Seg-1 to Seg-10 was purified by cutting agarose gel recovery and cloned into the pTOPO-TA vector supplied with the Zero Background pTOPO-TA Cloning Kit® (Aidlab, Beijing, China). Recombinant plasmid vectors containing inserts were transformed into Escherichia coli DH5α competent cells. Positive clones were identified by PCR using M13 universal primers, and sequenced using an automated ABI 3730 DNA sequencer (Applied Biosystems).
Sequence analysis and phylogenetic constructions
Initial sequence assembly and analyses were conducted using the Lasergene of DNAStar software package (Ver. 7.1.0; DNASTAR, Inc., Madison, WI). Sequences were identified by BLAST analysis (
All phylogenetic constructions and pairwise distance calculations were done using MEGA 6.0 with the p-distance parameter and 1000 bootstrap replicates (Tamura et al. 2013). GenBank nucleotide accession numbers for the sequences used for analysis and phylogenetic studies are listed in Supplementary Table S1.
Results
Mosquito collection
During an overnight, a total of 1150 mosquitoes were captured in one light trap in Mangshi City, Dehong Prefecture, Yunnan Province, China, on July 6, 2013. The mosquitoes belong to five species: Cx. tritaeniorhynchus Giles (n = 925, percent = 80.43%), Culex quinquefasciatus (n = 83, percent = 7.22%), Cx. pipiens pallens (n = 77, percent = 6.70%), Anopheles sinensis (n = 61, percent = 5.30%), and Armigeres subalbatus (n = 4, percent = 0.35%).
Virus isolation
A strain virus (DH13M98) was isolated from one pool of Cx. tritaeniorhynchus Giles. DH13M98 virus showed obvious CPE in c6/36 cells 72 h postinoculation in a second-blind passage. The CPE were characterized by strong refraction, spindle, rounding, rupture, cytolysis, and finally, all cells shedding (Fig. 2). However, no CPE were observed in BHK-21 cells through three blind passages.

Cytopathic effect of DH13M98 on C6/36 cells at 72 and 120 h ( × 100). (
Genomic dsRNA electropherotype
AGE analysis demonstrated that the genome of DH13M98 virus consists of 10 dsRNA segments. Exception of Seg-4 and Seg-5, which comigrate in this gel system, other segments migrate separately, forming a “1-1-1-2-1-1-2-1” pattern (Fig. 3), which is similar to that observed in UMAV (USA1969/01) and KHV (11RS20).
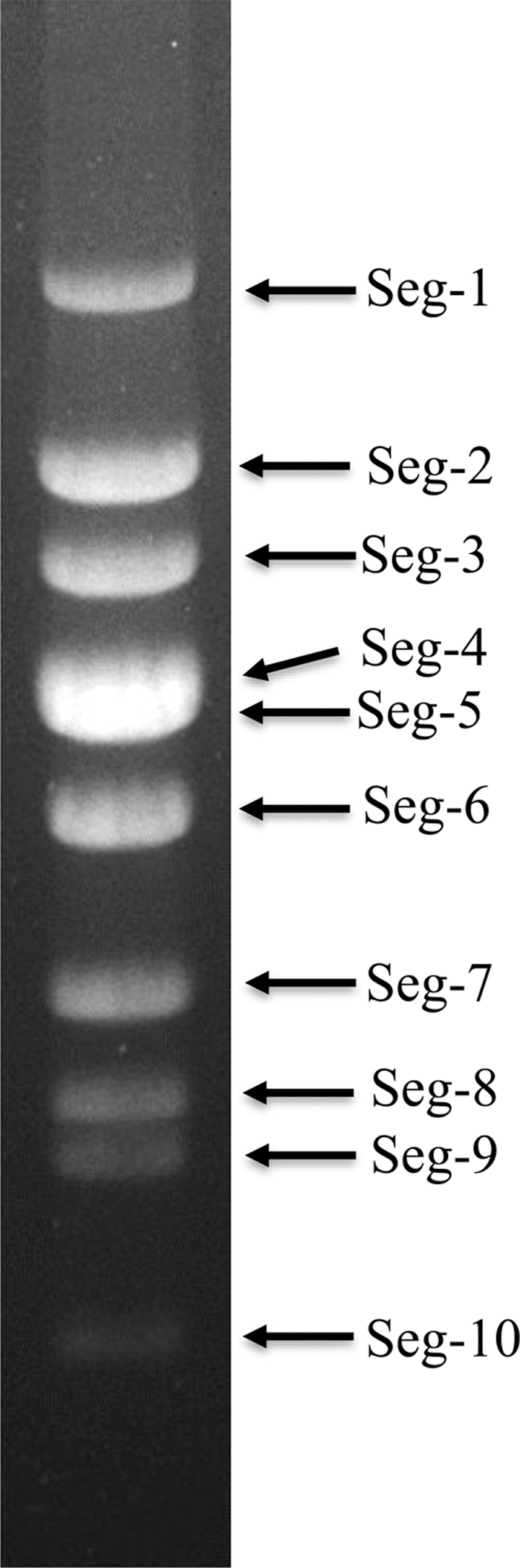
Agarose gel (1%) electrophoretic profile of dsRNAs of strain of DH13M98. dsRNA, double-stranded RNA.
Sequence analysis
Sequences of Seg-1 to Seg-10 of the DH13M98 virus have been deposited in the GenBank with accession numbers OM475538 to OM475547, respectively. The length of 1–10 segments varies from 3933 bp (seg-1) to 883 bp (seg-10) and their corresponding encoding proteins are presented in Table 1. Ten segments of the DH13M98 virus all share four fully conserved nucleotides at their 5′ ends, and five at their 3′ ends (5′-GUUU……GAUAC-3′). DH13M98 is similar to other orbiviruses with two terminal nucleotides at the 5′ end and three nucleotides at the 3′ end (5′-GU … UAC-3′), is conserved, and the first and last two nucleotides of each segment are reverse compliments. The 5′and 3′ noncoding regions (NCRs) of DH13M98 comprised 4.973% of the total genome, and the GC content of it was 43.71%.
Lengths of Double-Stranded RNA Segments 1–10, Encoded Putative Proteins, 5′ and 3′ Noncoding Regions of DH13M98 Virus Genome
aa, Amino acid; CaP, minor core protein-capping enzyme; Hel, minor core protein-helicase enzyme; NCR, noncoding region; NS, nonstructural proteins; OC1, outer capsid protein one; OC2, outer capsid protein two; Pol, RNA-dependent RNA polymerase; T13, major core-surface; T2, major subcore-shell protein; TuP, tubule protein; Vip, viral inclusion body protein; VRP, virus release protein.
Phylogenetic analysis of VP1 (Pol) proteins
Pol protein, encoded by Seg-1 of orbivirus, is very conservative and has previously been used in phylogenetic studies to classify new virus isolates at both the species and genus levels (Shaw et al. 2007). The aa sequence analysis of the VP1 (Pol) protein showed that DH13M98 shared 49.7–57.5% aa identity to VP1(Pol) of other MBOs; 46.1–48.2% aa identity to the CBOs; 47.8–50.3% to the TBOs; and only 37.5% to St Croix River virus (SCRV). However, DH13M98 shared highest levels of aa identity 89.5–98.7% with VP1 (Pol) of UMAV (IA08), UMAV (USA196901), SLOV, KHV, UMAV (ED-I-93-19), and UMAV (M4941_15), respectively (Table 2). Phylogenetic analysis based on Pol protein (Fig. 4) showed that all orbiviruses were formed from three major evolutionary branches: CBOs, MBOs and TBOs; in the evolutionary branch of MBOs, the DH13M98 virus and other UMAVs (including SLOV and KHV) form an independent evolutionary branch.
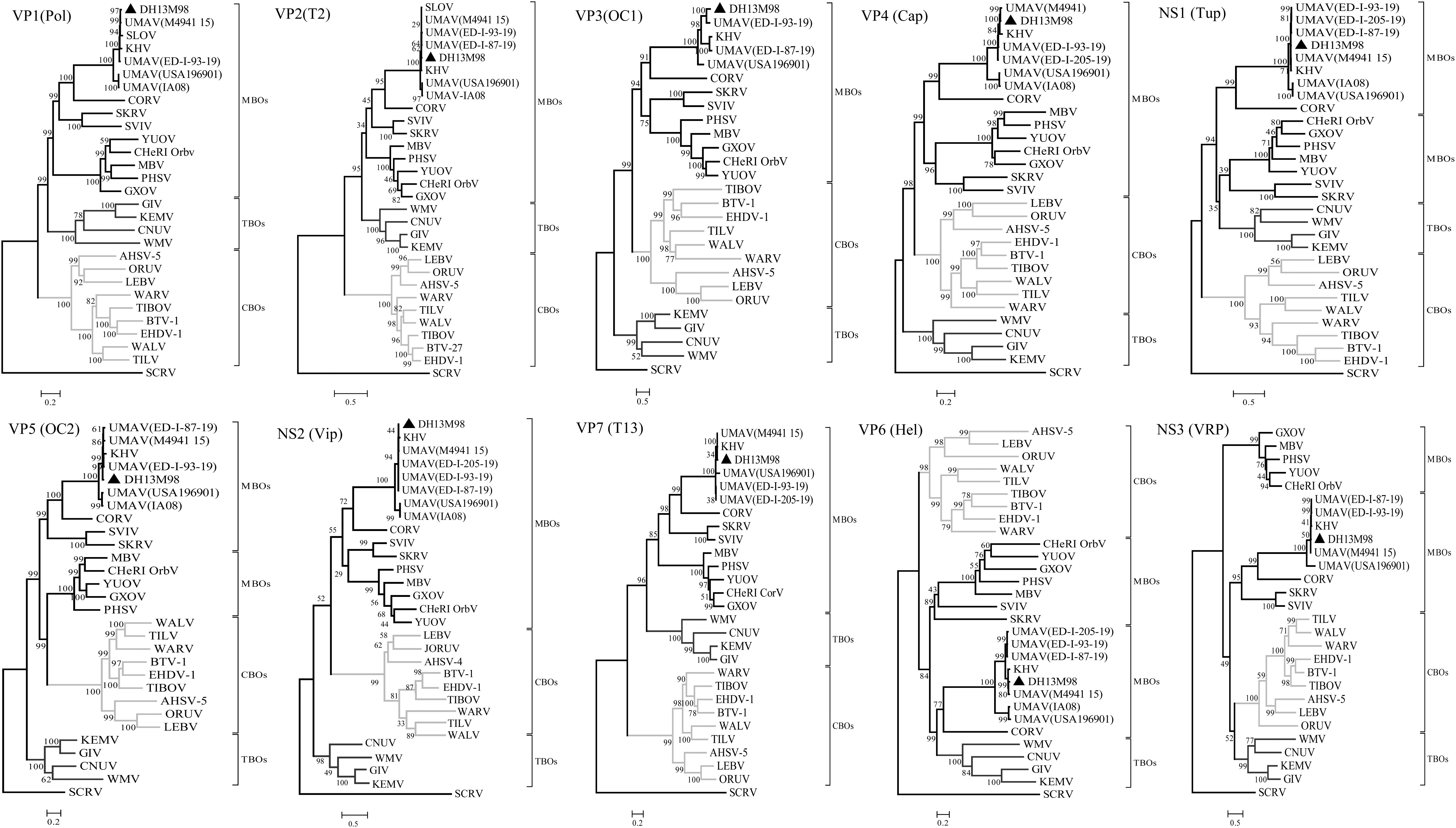
Phylogenetic trees of the coding regions of the 10 segments of DH13M98. Maximum-likelihood analysis in combination with 1000 bootstrap replicates was used to derive trees based on the predicted protein sequences. A scale representing the number of aa changes is shown in each panel. Bootstrap values are shown above branches to the left of major nodes. The DH13M98 isolates characterized in this study are marked with a black triangle. Full names of virus isolates and accession numbers are listed in Supplementary Table S1. aa, amino acid; CBOs, Culicoides-borne; MBOs, mosquito-borne; TBO, tick-borne.
Amino Acid Sequence Identity (%) Between DH13M98 and Other Orbiviruses
Arthropod vector: C, Culicoides; M, mosquito; T, tick. Full names of virus isolates and accession numbers are listed in Supplementary Table S1.
—, Not available; AHSV, African horse sickness virus; BTV, bluetongue virus; CNUV, Chenuda virus; CORV, Corriparta virus; EHDV, epizootic hemorrhagic disease virus; GIV, Great Island virus; GXOV, Guangxi orbivirus; KEMV, Kemerovo virus; KHV, Koyama Hill virus; ORUV, Orungo virus; PHSV, Peruvian horse sickness virus; SCRV, St Croix River virus; SKRV, Skunk River virus; SLOV, Stretch Lagoon orbivirus; UMAV, Umatilla virus; WARV, Warrego virus; WMV, Wad Medani virus.
Phylogenetic analysis of “T2” proteins
Orbiviral T2 protein phylogeny often correlates with the arthropod vector and can be classified into two groups: those encoded by Seg-2 (MBOs and TBOs) and those encoded by Seg-3 (CBOs) (Belaganahalli et al. 2015). The T2 protein of the DH13M98 is encoded by Seg-2. The T2 protein of DH13M98 and UMAV (including SLOV and KHV) shares 95.0–99.3% aa identities (Table 2), which are above the T2 aa identity threshold of 83% used for species determination by Attoui et al. (2005). Comparisons with other orbiviruses showed a maximum of 47.8% aa identities with Corriparta virus (CORV), down to 21.8% with SCRV. Phylogenetic analysis based on T2 protein (Fig. 4) showed that the DH13M98 and UMAV clustered together within the MBO group.
Phylogenetic analysis of outer-core “T13” proteins
The core surface protein VP7(T13) is a strongly immunodominant serogroup-specific antigen and is highly conserved within each Orbivirus species (Gumm and Newman 1982). The aa sequence analysis of the VP7 (T13) protein showed that DH13M98 shared the highest aa identity levels of 99.4% to UMAV (M4941_15), 99.1% to KHV, 96.2% to UMAV (IA08, ED-I-205-19 and ED-I-93-19), 91.1% to UMAV (USA196901), and only shared 29.4–42.2% aa identity with those of other MBO orbiviruses such as Guangxi orbivirus (GXOV) and CORV, but shared lower aa identities with the CBO orbiviruses such as 20.7% to Orungo virus (ORUV) and 25.2% to EHDV, or the TBO orbiviruses such as 26.8% to Great Island virus (GIV) and 28.3% to Chenuda virus (CNUV), but only 16.8% aa identity with the more distantly related SCRV (Table 2).
Phylogenetic analysis based on VP7(T13) protein exhibited similar topology to the T2 and Pol trees with three distinct “vector-groups” (CBOs, MBOs, and TBOs), meanwhile, DH13M98 and UMAV again clustered in an independent branch within the MBO group (Fig. 4).
Phylogenetic analysis of OC1
OC1 determines the Orbivirus serotype and is highly variable in both its aa sequence and size. OC1 is encoded by Seg-2 (VP2) in the CBOs, by Seg-3 (VP3) in the MBOs, and by Seg-4 (VP4) in the TBOs (Belhouchet et al. 2010). In DH13M98 and UMAV, VP2 (OC1) is encoded by Seg-3, consistent with mosquito transmission. The aa sequence analysis of the VP2 (OC1) protein showed that DH13M98 shares 79.2%, 60.7%, 57.4%, and 57.1% aa identity with UMAV (ED-I-93-19, USA196901, and ED-I-87-19) and KHV in OC1 protein, respectively; shares 10.8–13.3% aa identity with those of other MBO orbiviruses such as GXOV and Skunk River virus (SKRV), shares 6.9–9.8% aa identity with Warrego virus (WARV) and EHDV within CBO orbiviruses, shares 8.4–11.0% aa identity with Wad Medani virus (WMV) and Kemerovo virus (KEMV) within TBO orbiviruses (Table 2).
Phylogenetic analysis based on OC1 showed three major clusters that again correspond to their “vector groups” in a manner similar to the trees for Pol, T2, and T13 proteins (Fig. 4).
Phylogenetic analysis of other structural and nonstructural proteins
The aa sequence analysis of other structural and nonstructural proteins showed that DH13M98 shares 79.4–98.6% aa identity in Cap, 87.9–99.3% in Tup, 89.0–99.3% in OC2, 76.1–99.2% in Vip, 66.2–97.3% in Hel, and 66.3–98.8% in virus release protein (VRP) in relation to UMAV, but shares ≤48.4% aa identity in Cap, ≤29.5% in Tup, ≤47.4% in OC2, ≤29.9% in Vip, ≤26.5% in Hel, and ≤20.4% in VRP in relation to other orbiviruses (Table 2). Phylogenetic analysis based on those proteins of DH13M98 and other orbiviruses showed that the DH13M98 and UMAV are more closely related to each other than other orbiviruses in Cap, Tup, OC2, Vip, Hel, and VRP, and that the Cap and Vip all show similar relationships to those seen in Pol, T2, and T13 proteins, with distinct monophyletic groups for the TBOs, MBOs, and CBOs (Fig. 4).
Discussion
Parameters recognized by the ICTV for the polythetic “definition” of individual Orbivirus species include the following: the reassortment of genome segments, genome segment migration patterns during AGE, conserved terminal nucleotide sequences, serological cross-reactions, comparison of homologous genome segments by sequence analysis or cross-hybridization, host and vector range, and the nature of the clinical signs induced (Mertens et al. 2005). In this study, some features of DH13M98 resembled the USA1969/01 strain of UMAV (Belaganahalli et al. 2011b) and the 11RS20 strain of KHV (Ejiri et al. 2014) in terms of genome segment migration patterns, and conserved terminal nucleotide sequences of the 5′ and 3′′ NCRs, suggesting that the DH13M98, UMAV and KHV may belong to the same species.
The Orbivirus polymerase “Pol,” subcore-shell “T2,” and outercore “T13,” proteins are all highly conserved, with >73%, >83%, and >73% aa identity, respectively, within a single Orbivirus species (Belaganahalli et al. 2013, Cooper et al. 2014). Consequently, these proteins and their comparisons have been used as “markers” for the identification and classification of both existing and novel Orbivirus isolates. DH13M98 share 89.5–97.9%, 95.0–99.3%, and 91.1–99.4% aa identity with among members of the UMAV in Pol, T2, and T13 proteins, respectively, beyond the threshold for virus species classification, indicating that DH13M98 belong to the UMAV species.
The aa sequence of the outer capsid protein “OC1” protein is more variable (within each Orbivirus species) than any of the other viral proteins and determines viral serotype (Shirafuji et al. 2017). DH13M98 shared only 79.2%, 60.7%, 57.4%, and 57.1% aa identity with UMAV (ED-I-93-19, USA196901, and ED-I-87-19) and KHV in OC1 protein, respectively. A pairwise alignment of OC1 from different serotypes of BTV, EHDV, and AHSV indicated variations in the ranges of 28.3–64%, 31.1–76.7%, and 46–52% aa, respectively (Huismans et al. 2004, Anthony et al. 2009, Maan et al. 2011). Based on these data, the DH13M98 virus may be different serotypes within UMAV or belong to a higher variant strain. However, this needs to be further verified by the serum neutralization test.
UMAV has been isolated from mosquitoes many times (Cowled et al. 2009, Belaganahalli et al. 2011b, Ejiri et al. 2014, Tangudu et al. 2019). In this study, the strain DH13M98 was also isolated from mosquitoes. The aa sequence analysis of all ten proteins of DH13M98 showed that DH13M98 share higher aa identities with the MBO than the CBO or TBO Orbivirus species. Previous studies have observed that the genomes of orbiviruses contain 4.874–5.695% of NCR in the MBO group, 4.47–4.9% of NCR in the TBO group, and 3.5–4.1% NCR in CBO viruses (Belaganahalli et al. 2011b, Kapoor et al. 2013). Analysis of the DH13M98 genome showed that the NCR comprises 4.973% of the genome, within the range of the CBO group.
In addition, the GC content was 43.71% in DH13M98, which was within the range for MBO Orbivirus species, from 36.7% in Peruvian horse sickness virus (PHSV) to 45.1% in CORV, significantly lower than that of the TBO or tick-associated orbiviruses (51.93% in SCRV and 57.29% in GIV) (Belaganahalli et al. 2013, Kapoor et al. 2013). These data suggested that mosquitoes may be biological vectors of the DH13M98 virus.
In this study, the DH13M98 was isolated from Cx. tritaeniorhynchus Giles that was collected at a bovine shelter in the rural of Mangshi City. The mosquito was widely distributed in China, and a variety of human and animal viruses were isolated from it (Liang et al. 2018). In addition, it is the most important vector of the Japanese encephalitis virus in China (Liu et al. 2018). However, it is currently unknown whether the DH13M98 can infect either humans or animals. To determine whether this virus poses a risk to public health, an extensive serological study to define potential human and animal exposures to the DH13M98 is needed.
This is the first report of isolation of UMAV from mosquitoes in the southwest border area of Yunnan Province of China. These findings and the sequence data of DH13M98 will not only help to expand our knowledge of Orbivirus evolution and distribution, but also can be referenced to design UMAV diagnostic assays and facilitate further UMAV epidemiological studies in China.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors. Authorization for the collection of mosquitoes has been obtained from Institute for Yunnan Animal Science and Veterinary Institute, Kunming, China.
Footnotes
Acknowledgments
We would like to thank Professor Wu Aiping and Liu Lin at the Center for Systems Medicine, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences & Peking Union Medical College for the next-generation sequencing date analysis. We acknowledge the cooperation and enthusiasm of the Animal Disease Prevention and Control centers in Dehong prefecture. We are grateful to researcher Huachun Li for his financial support in the process of sample collection.
Author Disclosure Statement
The authors have declared that no competing interests exist.
Funding Information
This study was funded by Basic Research Projects of Yunnan Province (2019FA015, 202201AS070062); Yunnan Science and Technology Talents and Platform Plan (2018HB046); Yunnan Chenggong expert workstation (202005AF150034); projects funded by the central government to guide local scientific and Technological Development (202207AB110006); and independent research project of Yunnan Tropical and Subtropical Animal Viral Disease Laboratory (2022RW001).
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.