
Abstract
Background:
Ticks are an important vector among arthropods associated with serious medical and veterinary problems. In this research, we investigate Borrelia species in ticks isolated from the surface of livestock (sheep and goat) in different regions of West Azerbaijan province.
Materials and Methods:
Polymerase chain reaction (PCR) was performed using specific primers targeting Borrelia spp. genes. Borrelia spp. was identified through PCR. The positive PCR products were sent to Pishgam Company for sequencing. Sequenced data were analyzed, and phylogenetic analysis was performed using maximum likelihood method in MEGA V.10.
Results:
The detection rate of Borrelia spp. for 16srRNA gene 69 (n = 542; 12.7%; 95%Cl: 10.1%–15.8%), 42 (n = 542; 7.7%; 95%Cl: 5.78%–10.1%) positive on 5S-23SrRNA gene and ospA gene 4 (n = 542; 0.74%; 95%Cl: 0.29%–1.88%).
Conclusion:
These results are the first report in Iran to identify Borrelia spp. These results of the study showed that the Borrelia spp. in hard ticks detected by PCR and it was negative to soft ticks. it is important in public health implications at the studied areas.
Introduction
Ticks, as crucial vectors, transmit pathogens, such as bacteria, protozoa, and viruses, threatening human and animal health (Uilenberg et al., 2004). As Gram-negative bacteria, Borrelia spp. has a spiral form and belongs to the Spirochaetaceae class (Parola and Raoult, 2001). The bacteria are arthropod-borne diseases pathogens creating challenges for public health. Researchers have divided Borrelia bacteria's phylogenetic trees into three-categories: reptile-associated (REP), relapsing fever (RF), and Lyme borreliosis/Lyme disease (Takano et al., 2010). Etiological factors of Lyme borreliosis are Borrelia garinii, Borrelia burgdorferi sensu stricto, and Borrelia afzelii. Hard ticks of different species of Ixodes (e.g., Ixodes persulcatus, Ixodes scapularis, Ixodes ricinus) transmit them (Steere et al., 2016). RF borreliae's etiological factors include more than 20 species, such as Borrelia duttonii in Africa and Borrelia hermsii in North America. The major vectors of RF borreliae include soft ticks of the genus Ornithodoros (Wang et al., 2015).
Nevertheless, part of RF borreliae is spread by hard ticks such as Borrelia miyamotoi (Ixodes), Borrelia theileri (Rhipicephalus), and Borrelia lonestari (Amblyomma) (Wang et al., 2015). Soft tick-borne relapsing fever (STBRF) is highly prevalent in most regions of Iran (Asl and Nateghian, 2009; Karimi-Nejad, 1981; Naddaf et al., 2015). According to molecular characterization analyses and animal pathogenicity, different species of RF Borrelia in Ornithodoros soft ticks exist in Iran (Asl and Nateghian, 2009; Karimi-Nejad, 1981; Naddaf et al., 2012; Rafinejad et al., 2011). Borrelia spp. is spread by Ornithodoros tholozani and is the main source of STBRF in Iran (Karimi-Nejad, 1981; Naddaf et al., 2012). In areas without the fspi occurrence of O. tholozani, molecular and epidemiological evidence implies Borrelia microti (Asl and Nateghian, 2009; Naddaf et al., 2012), and strictly related species, the supposed ecotypes of B. duttonii, as the disease source (Naddaf et al., 2020; Naddaf et al., 2015). As the main arthropod-vectors, ticks are related to considerable veterinary and medical health challenges (Colwell et al., 2011).
Recently, there have been increasing concerns about the global climate change impact on the appearance of zoonotic diseases (Dietrich et al., 2011). Remarkably, climate change has resulted in several tick-borne diseases. The diseases are commonly observed in veterinary medical clinical settings, suggesting the necessity for a one-health approach. The genus Borreliacea is a group of motile bacteria with a helical shape that constitutes a monophyletic lineage within the phylum Spirochetes and includes two main clades. Previous studies hypothesized the association between certain bacteria groups and specific species of tick vectors; however, it is controversial.
Soft ticks of the genus Argas Latreille, 1795 (Ixodida; Argasidae) have a global distribution and contain ∼60 species (Palomar et al., 2021). Only eight of them are defined in Western Palearctic area, definitely, Argas persicus, Argas gilcolladoi, Argas vespertilionis, Argas reflexus, Argas transgariepinus, Argas polonicus, Argas vulgaris, and Argas macrostigmatus (Dantas-Torres et al., 2009; González-Acuña and Guglielmone, 2005; Palomar et al., 2021). Almost all of them were identified as parasites of bats or birds in Spain (Southwestern Europe) (Palomar et al., 2021; Reeves and Lloyd, 2019).
The genus Argas contains species that transmit medical and veterinary pathogens. In addition to conditions directly created by soft ticks (e.g., anaphylaxis and toxicosis) (Kazim et al., 2021; Sarwar, 2017), microorganisms are carried by the ticks that can be infectious problem agents. Explicitly, Argas species can vector bacterial pathogens, like Aegyptianella spp. and Borrelia anserine, as well as viruses like the Issyk-kul (Palomar et al., 2021; Sili, 2017). Argas ticks detect other microorganisms with unproved pathogenicity: bacteria from genera Borrelia, Anaplasma, Ehrlichia, Bartonella, Francisella, Rickettsia, Coxiella, Rickettsiella, and protozoans like Hemolivia spp. and Babesia, and viruses that belong to Orthomyxoviridae, Flaviviridae, Orthonairoviridae, Phenuiviridae, and Reoviridae families (Dantas-Torres, 2008; Ebani and Mancianti, 2022).
Materials and Methods
Study areas
West Azerbaijan is a province in northwest Iran with different geographical regions, such as flat areas and mountainous regions, as well as the Lake Urmia coastline. This province's climate is principally influenced by the Atlantic Ocean and Mediterranean rainy winds (Fig. 1) (Khademi et al., 2020).
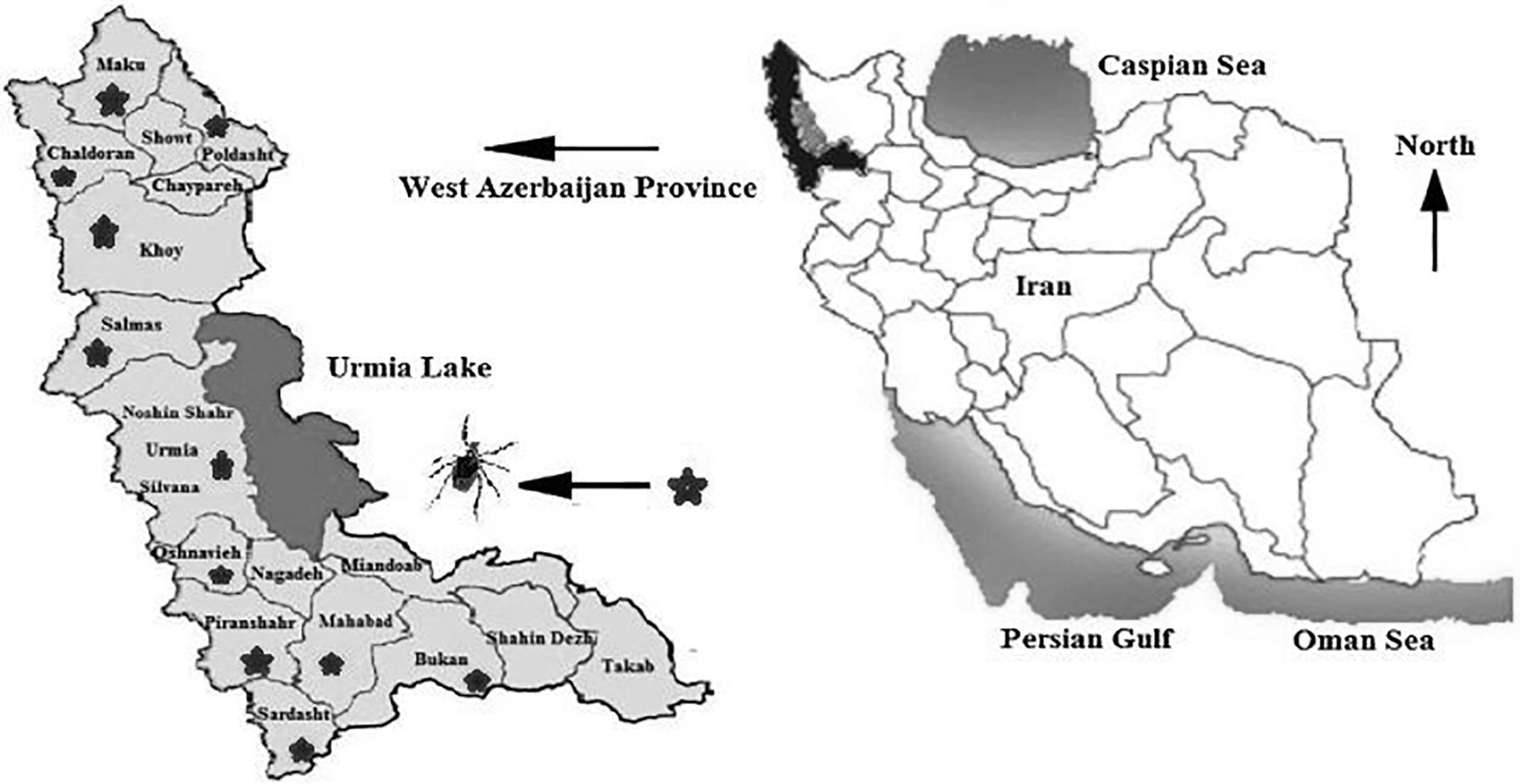
Schematic map of the study areas, West Azerbaijan, Iran.
Sample collection
The sampling process was conducted in various regions of West Azerbaijan province, consisting of three geographical areas (north, center, and south). Tick samples were collected from the surface of sheep and goats by securing them with the assistance of individuals and removing ticks using tweezers. Ticks were gathered from various body parts of goats and sheep in the west Azerbaijan province throughout the spring and summer of 2022. To identify the tick species, individual samples were collected in sterile glass bottles containing 95% ethanol. Afterward, these samples were conveyed to the Parasitology Laboratory at the Urmia University's Faculty of Veterinary Medicine. At the laboratory, a loupe microscope and reliable diagnostic keys were utilized to identify the ticks. To detect bacterial species, the bacteriology laboratory utilized the nested-PCR method, examining the 16S rRNA, 5S-23S rRNA, and ospA genes (Walker, 2003).
DNA extraction
To eliminate any alcohol residues, the ticks were washed twice with phosphate-buffered saline before being fixed in liquid nitrogen. Subsequently, the ticks were crushed in a sterile environment using a scalpel blade and transferred into an ependorff microtube for the extraction process. Then they were moved for DNA extraction following their removal from 70% ethanol, after which ticks were dehydrated on clean paper using airflow. To extract DNA, a DNA Extraction Kit from MBST was employed. This kit, commercially available, facilitated the extraction process (Medlock et al., 2018). The quantity and quality of the extracted DNA were assessed by NanoDrop 2000c (Thermo Scientific). The extracted DNA was kept at −20°C for subsequent use in PCR. The negative control of extraction was the elution buffer from the extraction kit during the DNA extraction process.
Molecular detection of Borrelia spp.
The DNA was tested for the presence of the specific bacteria using conventional nested PCRs. The target genes included the 16S rRNA as well as the intergenic 5S-23S rRNA and ospA genes for Borrelia spp. Primer sets were designed from Amplifx (Version 1.7.0) software, as shown in Table 1. For primer design, Borrelia gene sequences were selected from the National Center for Biotechnology Information (NCBI) website, for the 16S rRNA gene from the sequence with the GenBank number CP086555, for the 5S-23S rRNA genes from the GenBank sequence number CP088943, and for the design of the ospA gene, accession number MN461278 was used. The PCR reaction was performed in a 25 μL volume, consisting of 4 μL of template DNA (50–100 ng/μL), 1 μL of each primer (concentration of primer 0.1 nmol/μL), and 12.5 μL of master mix, and the total volume was completed with sterile distilled water. A touchdown PCR program was used (Table 1) and performed in the thermal cycler (Quanta Biotech) to minimize nonspecific amplification. The touchdown PCR program typically involves an initial high annealing temperature, gradually decreasing in subsequent cycles, promoting specific amplification while reducing nonspecific amplification. For the nested PCR, 2.5 μL of the initial PCR product was used as a template for the second round of amplification. The PCR products were then subjected to electrophoresis on a 2% agarose gel containing a safe stain (Labnet; ENDURO). The gel was subsequently visualized using the Genius Gel Documentation system (Figs. 2 –4) (Syngene Bio-Imaging) (Reifenberger et al., 2022).

Agarose gel electrophoresis image of nested PCR test on ticks to identify Borrelia spp., with 16S rRNA gene (597 bp), which had (Lane M-marker 50 bp DNA [Smobio Technology, Inc.]. Lanes 1–4 positive tick samples, N Lane of negative control).
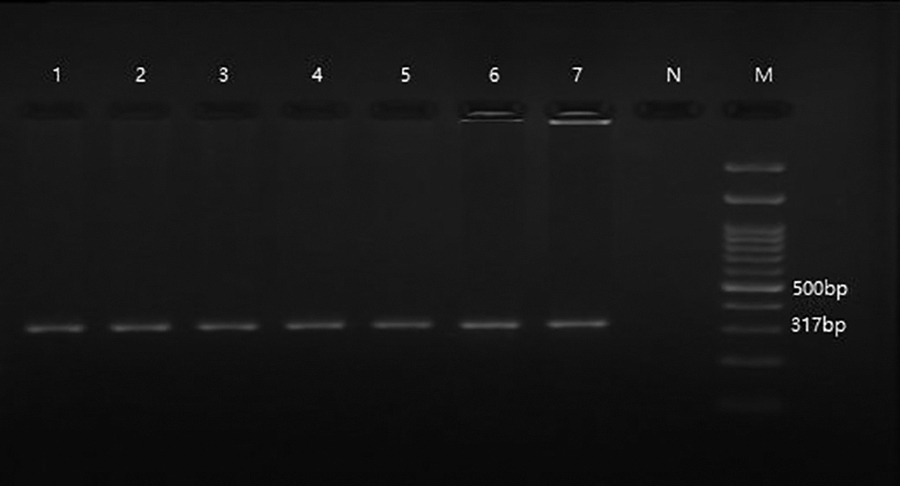
Agarose gel electrophoresis image of nested PCR test on ticks to identify Borrelia spp., with 5S-23S rRNA gene (317 bp), which had (Lane M-marker 100 bp DNA [Smobio Technology, Inc.]. Lanes 1–7 positive Tick samples, N Lane of negative control).

Agarose gel electrophoresis image of nested PCR test on ticks to identify Borrelia spp., with ospA gene (252 bp), which had (Lane 6-marker 50 bp DNA [Smobio Technology, Inc.]. Lanes 1–4 positive Tick samples, 5 Lane of negative control).
Primer Sequences for Detection of Borrelia spp. Gene by Using the Nested PCR Method
Nucleotide diversity and phylogenetic tree construction
Some positive samples were sent to Pishgam Biotechnology Company in Tehran for sequencing using the Sanger method. The obtained sequences were then uploaded to the NCBI website for analysis and blasting. For drawing the phylogeny tree (Figs. 5 –7), 596 nucleotides were from the 16S rRNA gene, 317 nucleotides from the 5S-23S rRNA gene, and for the ospA gene, 252 nucleotides were used. Sequences were uploaded to the NCBI to seek the most analogous reference sequences; furthermore, the Country of Origin Information (COI) positions were identified using NCBI's BLAST. For phylogenetic analysis, all Borrelia spp. COI sequences accessible in the GenBank were utilized. Using the Clustal W alignment program, the alignment was manually adjusted to eliminate any related errors before being released as MEGA and FASTA files (Paulo and Pappas, 2018). All acquired nucleotide sequences were entered into the GenBank and given accession numbers. The phylogenetic association was investigated and developed using the Molecular Evolutionary Genetics Analysis software version 10 by the maximum-likelihood method. One thousand bootstraps were used to test the reliability of an inferred tree. MEGA version 10 and BLASTN software were used to analyze DNA sequence polymorphism to assess nucleotide diversity (Yarza et al., 2008).

The phylogenetic tree of Borrelia spp. 16S rRNA gene sequences were gained and put in GenBank with various accession numbers. Bold arrows shows the 16SrRNA gene sequences gained in the present work. The neighbor-joining approach of MEGA 10 was used to infer the tree. Branch points show bootstrap values. The numbers above branches indicate bootstrap support of 1000 replicates.

The phylogenetic tree of Borrelia spp. 5S-23S rRNA gene sequences were gained and put in GenBank with various accession numbers. Bold arrows shows the 5S-23S rRNA gene sequences gained in the present work. The neighbor-joining approach of MEGA 10 was used to infer the tree. Branch points show bootstrap values. The numbers above branches indicate bootstrap support of 1000 replicates.

The phylogenetic tree of Borrelia spp. ospA gene sequences were gained and put in GenBank with various accession numbers. Bold arrow shows the ospA gene sequences gained in the present work. The neighbor-joining approach of MEGA 10 was used to infer the tree. Branch points show bootstrap values. The numbers above branches indicate bootstrap support of 1000 replicates.
Data analysis
The chi-square test was used for statistical data analysis, and SPSS software Version 22 was employed. The p values below 0.05 were regarded as significant.
Results
The positive rate of Borrelia spp. DNA was 12.73% and 7.74% for tick's samples using 16S rRNA and 5S-23SrRNA genes, respectively. The Borrelia spp. was identified based on 16SrRNA and 5S-23S rRNA genes under the accession numbers OQ073805.1, OR342388.1 and OR352151.1, OR345451.1. Furthermore, the findings regarding the molecular prevalence of Borrelia spp. DNA indicated that out of 542 samples, 69 (12.7%; 95% confidence interval [CI]: 10.1–15.8%) were tested positive for the 16S rRNA gene, while 42 (7.7%; 95% CI: 5.78–10.1%) were positive for the 5S-23S rRNA gene (Table 2).
Prevalence of Borrelia spp. in Ticks Samples from West Azerbaijan in Iran Assessed by the Nested PCR
One of the four detected ospA gene sequences that resulted in this study was recorded in NCBI with accession number OQ341518, 4 (n = 542; 0.74%; 95% CI: 0.29–1.88%) positive for the prevalence of Borrelia spp. DNA (Table 2). This study focused on two species of hard ticks (Hyalomma and Ripicephalus) and one species of soft tick (Argas reflexus). The ticks were categorized into male and female groups using diagnostic keys and then analyzed separately based on gender and tick type using the PCR method. The results showed that female ticks had the highest Borrelia infection rate at 14.30%, while male ticks had a lower infection rate of 11.70%.
In Hyalomma ticks, prevalence of Borrelia was higher than Ripicephalus ticks. It is crucial to note that none of the soft tick samples tested positive for Borrelia bacteria.
Discussion
Hard ticks (Ixodidae) transfer various pathogenic veterinary and medical agents. In recent decades, the number of tick-borne cases and the abundance of ticks have increased in many European countries (Baneth, 2014; Körner et al., 2021). Lyme disease, or Lyme borreliosis, has the highest prevalence as the human tick-borne infection in the north of the equator, and its incidence has exhibited an increase in at least nine countries in Europe (Blazhev et al., 2022; Stone et al., 2017).
Based on 16SrDNA sequences, indicated the phylogenetic associations of RF-related Borrelia species. They showed that Borrelia recurrentis and B. duttonii were from the same group, and B. hermsii was contained in another group shared by other Borrelia species such as Borrelia turicatae and Borrelia parkeri. The spirochaete separated from Hyalomma aegyptium are grouped between Lyme borreliosis agents and RF-associated Borrelia species. RF-associated Borrelia species are transferred primarily by soft ticks. However, hard ticks, that is, Amblyomma americanum in the United States and I. persulcatus in Japan carry two RF-associated Borrelia species, B. lonestari’, and B. miyamotoi (Fukunaga et al., 1995; Morel et al., 2019).
The Ixodes species of hard ticks transmit Lyme-disease-related Borrelia species. Nevertheless, previous studies reported the Borrelia detection by PCR from Hyalomma marginatum hard ticks gathered in Portugal, specified as Lymeborreliosis-related Borrelia lusitaniae (De Michelis et al., 2000). Rickettsia mongolotimonae and Crimean-Congo hemorrhagic fever virus are other pathogens transmitted by the members of the genus Hyalomma (Parola and Raoult, 2001). It is the first case of the separation of a Borrelia spp. in Iran.
According to the present research, Borrelia species were found in 12.73% of West Azerbaijan hard ticks and 0% of A. reflexus soft ticks. The ticks were from the genera Rhipicephalus sanguineus, Hyalomma aegyptium, Hyalomma anatolicum, Hyalomma asiaticum, and the A. reflexus, and Rhipicephalus turanicus genera did not have any Borrelia species (Table 2).
None of the 104 tick samples verified by Cotes-Perdomo et al. (2022) was infected with any Borrelia species, and ticks were isolated by gender. Despite the positive test of hard ticks in the current work for Borrelia species, male and female ticks did not significantly differ in infection levels (Cotes-Perdomo et al., 2022).
The highest infection rate was found in female ticks (43.7%); however, this difference was not statistically significant, according to one study by Blazhev et al. (2022) in Bulgaria. Our findings were consistent with those of this investigation (Blazhev et al., 2022).
McCoy et al. (2014) used the nested PCR technique to identify Borrelia species, and their research in Mali confirmed that the ticks were infected with the Borrelia species.
Considering the results of nested PCR, 542 ticks samples were investigated with primers targeting the 16SrRNA and 5S-23SrRNA genes, represented 130 soft ticks samples (0%), 412 hard ticks samples with primers targeting the 16SrRNA and 5S-23SrRNA genes positive samples of 69 (12.73%), 42 (7.74%), and ospA 4 (0.73%) were revealed from regions of West Azerbaijan province in Iran. Obviously, the Borrelia spp. infection molecular prevalence was reported in ticks for the first time in this study in Iran. This study aimed to comprehend the role of ticks (hard and soft) as potential spreaders and carriers of Lyme disease in Iran. PCR assay and sequencing were used to assess the exposure of ticks to Borrelia spp. The sequences of 5S-23S rRNA, 16S rRNA, and ospA genes acquired from positive DNA samples were analyzed, and it was agreed that they act as hosts.
B. garinii with OspA gene was the second most frequent strain in unfed nymphal ticks. The results of this study suggest the presence of specific transmission cycles involving larval and nymph I. ricinus ticks, B. burgdorferi, and B. garinii OspA type 4, potentially involving an unknown vertebrate host. Additionally, the detection of mixed infections in nymphs supports the possibility of a single-host species acting as a reservoir for both B. burgdorferi and B. garinii OspA types. I. ricinus ticks typically acquire borrelia infections during bloodmeals from infected hosts and predominantly transmit the infection trans-stadially, with occasional trans-ovarial transmission (Monin et al., 1989). Mixed infections in ticks may occur through feeding on different infected hosts or a single host simultaneously infected with multiple B. burgdorferi types. Cofeeding transmission between ticks on the same host could also lead to multiple infections in individual ticks (Gern and Rais, 1996).
Given that questing nymphs typically feed as larvae on a single host and trans-ovarial transmission is uncommon, the presence of mixed infections in unfed nymphs suggests either an infected host with multiple borrelia types or cofeeding transmission. These findings indicate the presence of one or more host species that serve as the primary hosts for I. ricinus larvae, filtering and favoring B. burgdorferi and B. garinii OspA. The study sites exhibited a diverse range of B. burgdorferi species and OspA types, emphasizing that for test and vaccine development in Europe, none of these species or OspA types can be disregarded. Additional evidence supports the existence of another transmission cycle that favors B. garinii OspA type 4, which is relatively rare in Europe (Lenčáková et al., 2006).
To clarify the genetic identity of Borrelia spp. spirochetes, their differential reactivity can be considered with PCR primers specific for genospecies targeting the 5S-23SrRNA intergenic spacer amplicon gene. It is essential to further classify genetic heterogeneity by analyzing longer sequence data within Borrelia spp. identified previously as the same genospecies of uncharacteristic strains of Borrelia spirochetes (Chao et al., 2013; Lohr et al., 2018; Ružić-Sabljić and Cerar, 2017). Furthermore, the 16SrRNA gene was found at a greater positive rate in ticks studied for Borrelia spirochetes.
According to a recent case report by Trevisan et al. (2022), 11 pregnant women were diagnosed with Lyme disease. These patients had a history of coming into contact with ticks and being bitten by them. Previous studies conducted in Iran and other parts of the world have highlighted the public health concern posed by diseases transmitted by ticks. For instance, Aqiqi et al. discovered that various tick species, such as O. tholozani, Ornithodoros canestrini, Ornithodoros lahorensis, Ornithodoros erraticus, A. persicus, and A. reflexus, are widely distributed in the Qazvin province of Iran (Trevisan et al., 2022).
In their research, it was discovered that O. tholozani was infected with B. persica, and O. erraticus was infected with B. microti. Another study conducted by Shirani et al. (2016) found that Borrelia persica was extracted from dog blood by soft oronthous ticks (Shirani et al., 2016). In a separate study by Naddaf et al. (2020), PCR amplification and sequencing of a 600 bp flaB sequence identified Borrelia bavariensis, B. garinii, B. afzelii, and B. valaisiana, as well as RF Borrelia and B. miyamotoi in I. ricinus ticks. The RF-like Borrelia found in Rhipiscephalus ticks showed the highest similarity (97.98%) to the strain infecting Hemophysalis megaspinosa ticks in Japan. Our analysis of phylogeny and BLAST indicates an expansion of the European Borrelia range associated with I. ricinus ticks in northern Iran (Naddaf et al., 2020).
Borrelia species were recognized, and it was found that Borrelia spp. was widely distributed in ticks in Iran. In this work, Borrelia spp. was found in ticks from West Azerbaijan Province. According to the phylogenetic and analysis sequences, those isolates were closely related to the consistent genotypes in terms of the 16SrRNA gene with higher sequence similarities (100%, GenBank acc. nos. OP293343).
A similarity of 100% was found in the 16SrRNA gene sequence of Borrelia spp. achieved from ticks to the gene of B. turcica in Turkey (GenBank acc. nos. AB111854, CP028884). B. turcica is related to the RF Borrelia spp. and is also based on the analysis of the 5S-23SrRNA gene, our gene sequence to GenBank acc. no. CP114738, which was related to B. miyamotoi in America. This is the first report regarding the agent of the Borrelia relapsing fever group in Iran, which is isolated from a tick and is caused by B. miyamotoi. In conclusion, infection with Borrelia spirochetes was successfully identified from tick samples from different district places in West Azerbaijan Province, Iran. Borrelia spp. was highly prevalent in the province's sampled areas. More studies are required on the occurrence of Borrelia spp. groups for both relapsing fever and Lyme disease to prove the existence of these various Borrelia spp. in ticks in Iran. We found that Borrelia infection in ticks could indicate a potential concern for public health. Future studies can warrant further widespread and detailed control of tick populations and the screening for Borrelia in various hosts.
Conclusion
It is the first study related to the Borrelia spp. prevalence in hard and soft ticks obtained from West Azerbaijan's presence in Iran. In this work, the existence of important pathogenic microorganisms, particularly in veterinary medicine, was recorded. According to the results of the research, such ticks as A. reflexus and those from the genera R. sanguineus, H. aegyptium, H. anatolicum, H. asiaticum, and the A. reflexus, and R. turanicus may be in the transmission of borrelia in a vector. Principally because of its significance for public health in the Lyme group data, it can be utilized in future works to develop control mechanisms for ticks. The presented molecular instrument can be used for investigating the genetic variability in Borrelia spirochetes found in various reservoir hosts and vector ticks, facilitating the knowledge of the importance of genetic diversity related to the epidemiological characteristics of Borrelia spirochetes in West Azerbaijan, Iran.
Ethic Approval
In this study, there were no work done on animals, and the tick samples were taken from the surface of the animal body only.
Footnotes
Acknowledgments
We gratefully acknowledge the sponsorship provided by the Faculty of Veterinary Medicine, Urmia University. Thanks are also due to Mr. Kazemnia, and Dr. Khademi who gave us much valuable technical assistance in the early stages of this work.
Authors' Contributions
A.E.: Preparation of proposal, sample collection, conduction of practical works, and manuscript writing. A.O.: Helped in proposal preparation and practical works, conclusion, and data analysis. M.T.: Helped in parasitology section (identification of ticks) and data analysis.
Author Disclosure Statement
Authors mention that there is no conflict of interest in this study.
Funding Information
This research is the subject of PhD thesis of A.E. that was conducted by financial support of Urmia University.