
Abstract
Background:
Kadipiro virus (KDV) is a species of the new 12 segmented RNA virus grouped under the genus Seadornavirus within the Reoviridae family. It has previously been isolated or detected from mosquito, Odonata, and bat feces in Indonesia, China, and Denmark, respectively. Here, we describe the isolation and characterization of a viral strain from mosquitoes in Yunnan Province, China.
Methods:
Mosquitoes were collected overnight using light traps in Shizong county, on July 17, 2023. Virus was isolated from the mosquito homogenate and grown using baby hamster kidney and Aedes albopictus (C6/36) cells. Preliminary identification of the virus was performed by agarose gel electrophoresis (AGE). The full-genome sequences of the strain were determined by full-length amplification of cDNAs and sequenced using next-generation sequencing.
Results:
We isolated a viral strain (SZ_M48) from mosquitoes (Culex tritaeniorhynchus Giles) that caused cytopathogenic effects in C6/36 cells. AGE analysis indicated a genome consisting of 12 segments of double-stranded RNA that demonstrated a “6-5-1” pattern, similar to the migrating bands of KDV. Phylogenetic analysis based on the full-genome sequence revealed that SZ_M48 is more clustered with KDV isolates from Hubei and Shangdong in China than with Indonesian and Danish strains. The identity between SZ_M48 and SDKL1625 (Shandong, China) is slightly lower than that of QTM27331 (Hubei, China), and the identity with JKT-7075 (Indonesia) and 21164-6/M.dau/DK (Denmark) is the lowest.
Conclusion:
The full-genome sequence of the new KDV strain described in this study may be useful for surveillance of the evolutionary characteristics of KDVs. Moreover, these findings extend the knowledge about the genomic diversity, potential vectors, and the distribution of KDVs in China.
Introduction
A new 12
The prototype species of the genus, BAV, is considered to be an emerging pathogen that causes human viral encephalitis (Xu et al., 1990). Mice infected with BAV and KDV did not die or show severe clinical presentations, and produced protective immunity (Attoui et al., 2006). BAV can productively infect mosquito cells and replicate on the only mammalian cells (BSR cells), while KDV has only been grown successfully in mosquito cell lines (Lv et al., 2012; Zhai et al., 2010). In addition to replicating in mosquito cell lines, LNV can also replicate in mammalian cell lines and has been able to infect and kill mice in experimental settings (Attoui et al., 2006).
KDV (JKT-7075) was first isolated from Culex fuscocephalus in Java, Indonesia, in 1981 (Brown et al., 1993). In 2014, Carolyne et al. detected the viral nucleic acids of KDV in the plasma of some Kenyan adults with unexplained fever, but the results were possibly due to contamination and could not be verified (Ngoi et al., 2017; Ngoi et al., 2016). In 2015, Christina et al. used a nontargeted approach of next-generation sequencing (NGS) to obtain the full genome of KDV (21164-6/M.dau/DK) from bat fecal samples in Denmark, marking the first detection of the virus in Europe (Lazov et al., 2021). In 2019, Junna et al. obtained some nucleic acid sequences of KDV from rat samples by viral metagenomic analysis using publicly available data, but the reliability of these data could not be determined (Kawasaki et al., 2021).
In China, five strains of KDV were isolated from three genera of mosquitoes (Culex tritaeniorhynchus, Anopheles sinensis, and Armigeres subalbatus) in Yunnan province in 2005, and the results were verified by PCR (Sun et al., 2009b). In 2013, Shi et al. obtained the full genome of a KDV strain (QTM27331) from Odonata samples collected from Hubei Province by using deep transcriptome sequencing (Shi et al., 2016). In 2016, one strain of KDV (SDKL1625) was isolated from Anopheles sinensis mosquitoes collected in northeast Shandong province (Zhang et al., 2018). Subsequently, four KDV strains were isolated from Culex tritaeniorhynchus collected in southwest Shandong province in 2021 (Yunjiao et al., 2023). Here, we report a new KDV strain, SZ_M48, which was isolated from mosquitoes collected in Shizong county, Yunnan province, China in 2023, with a detailed description of its full-genome analyses and molecular properties, as well as phylogenetic relationships with members of Seadornavirus.
Materials and Methods
Sample collection
Mosquito samples were collected from caprine shelters at night using light traps (12 V, 300 mA; Wuhan Lucky Star Environmental Protection Tech. Co., Ltd., Hubei, China) in outside Wulong Village (104°17′28″ E, 24°38′53″ N, altitude 968 m) in Shizong county, Yunnan Province on July 17, 2023. Light traps were set overnight, spanning the period from sunset to sunrise. Captured mosquitoes were frozen at −20°C for 30 min, then classified and identified on a chilled plate based on morphologic characteristics under a stereoscopic microscope. Each 50 female mosquitoes of the same species were pooled in a cryogenic vial and stored in liquid nitrogen until virus isolation was performed.
Cell culture and virus isolation
Baby hamster Syrian kidney (BHK-21) and Aedes albopictus (C6/36) cells were used in this study for virus isolation. The cell culture was performed according to a method previously described by Wang et al. (2015). Specimens were then homogenized, centrifuged, and blind passaged three times on monolayers of BHK-21 and C6/36 cells in 24-well plates, as previously described (Yang et al., 2023b). The cells were observed daily (days 1–7 postinoculation) for cytopathic effects (CPEs). Supernatants of cell cultures showing CPE were collected and the isolates were subsequently plaque purified in C6/36 cells using agarose and neutral red, and one clone was then propagated and stored at −80°C for additional analysis.
Reverse transcript-quantitative polymerase chain reaction and genomic electropherotype analysis
Total RNA was then extracted using RNAiso Plus (TaKaRa, Dalian, China) according to the manufacturer's instructions. In accordance with previous studies, the group-specific RT-qPCR targeting Akabane virus (AKAV), BAV, Bluetongue Virus (BTV), Epizootic hemorrhagic disease viruses (EHDV), Palyam virus (PALV), and Tibet orbivirus (TIBOV) were used for preliminary virus identification (Duan et al., 2022; Hofmann et al., 2008). Separation of viral double-stranded RNA (dsRNA) from the total RNA by precipitating high-molecular-mass single-stranded RNA in 2 M LiCl, as described by Attoui et al. (Attoui et al., 2000). Six microliters of viral dsRNA were taken for electropherotype analysis, and the remaining RNA was stored at −80°C. Viral genomic segments were separated by 1% agarose gel electrophoresis (AGE) at 90 V for 4 h in 1 × Tris-Acetate EDTA buffer, stained with Goldview II (Solarbio, Beijing, China), and visualized under ultraviolet light.
Full-length complementary DNA amplification and NGS
Viral dsRNA was reverse-transcribed into complementary DNA (cDNA) using full-length amplification of cDNAs (FLAC) technique as previously reported (Maan et al., 2007; Potgieter et al., 2009). In brief, the 3′ ends of viral dsRNA were ligated to an “anchor-primer,” with a phosphorylated 5′ terminus, using T4 RNA ligase, followed by reverse transcription using a PrimeScriptTM II Reverse Transcriptase Kit (TaKaRa). The resulting cDNAs were amplified using a primer complementary to the “anchor-primer” by high-fidelity PrimeSTAR® GXL DNA Polymerase (TaKaRa). The amplicons were analyzed by 1% AGE and then sent to Magigen Company (Guangzhou, China) for NGS using an Illumina HiSeq 2000 system. Subsequently, reads preparation, reads/sequences checking, and de novo assembling for each genomic segment of the virus were performed using the Soapnuke (v2.0.5), BWA (v0.7.17), and Megahit (v1.1.2) software, respectively (Chen et al., 2018; Li et al., 2016).
Sequence analysis and phylogenetic constructions
Reference sequences of KDV strains and representative members of the Seadornavirus genus (BAV, BALV, LNV, and Mangshi virus) were downloaded from GenBank on December 1, 2023 and are listed in Supplementary Table S1. Open reading frames (ORFs) of viral genomic segments were identified and translated into amino acid sequences using ORF-finder Home-NCBI (https://www.ncbi.nlm.nih.gov/orffinder/). Sequences were identified by BLAST analysis, and then multiple alignments of consensus sequences were performed using CLUSTAL W method (version 2) (Higgins et al., 1996). Nucleotide and amino acid (nt/aa) identities calculations and phylogenetic tree constructions were conducted using BioEdit Sequence Alignment Editor (v7.0.9.0) and MEGA (v6.06) with neighbor-joining methods in a pairwise deletion, p-distance algorithm, and bootstrapped using 1000 replicates (Tamura et al., 2013).
Results
Mosquito collection
During an overnight, a total of 1487 mosquitoes were collected in Wulong Village, Shizong county, Yunnan Province, China, on July 17, 2023. The mosquitoes belong to six species: Anopheles sinensis (n = 673, percent = 45.26%), Culex tritaeniorhynchus Giles (n = 466, percent = 31.34%), Culex pipiens pallens (n = 158, percent = 10.63%), Culex quinquefasciatus (n = 111, percent = 7.46%), Armigeres subalbatus (n = 67, percent = 4.51%), and Anopheles minimus (n = 12, percent = 0.81%).
Virus isolation
A strain virus (SZ_M48) was isolated from one of 10 Culex tritaeniorhynchus Giles pools that is without blood meals. The virus was shown to cause clear CPEs in c6/36 cells at 72 h postinoculation in a second-blind passage, but no CPEs were observed in BHK-21 cells during three consecutive passages. The CPEs are characterized by strong cell shrinkage, shedding, cytolysis, and finally detachment from the growth surface (Fig. 1).
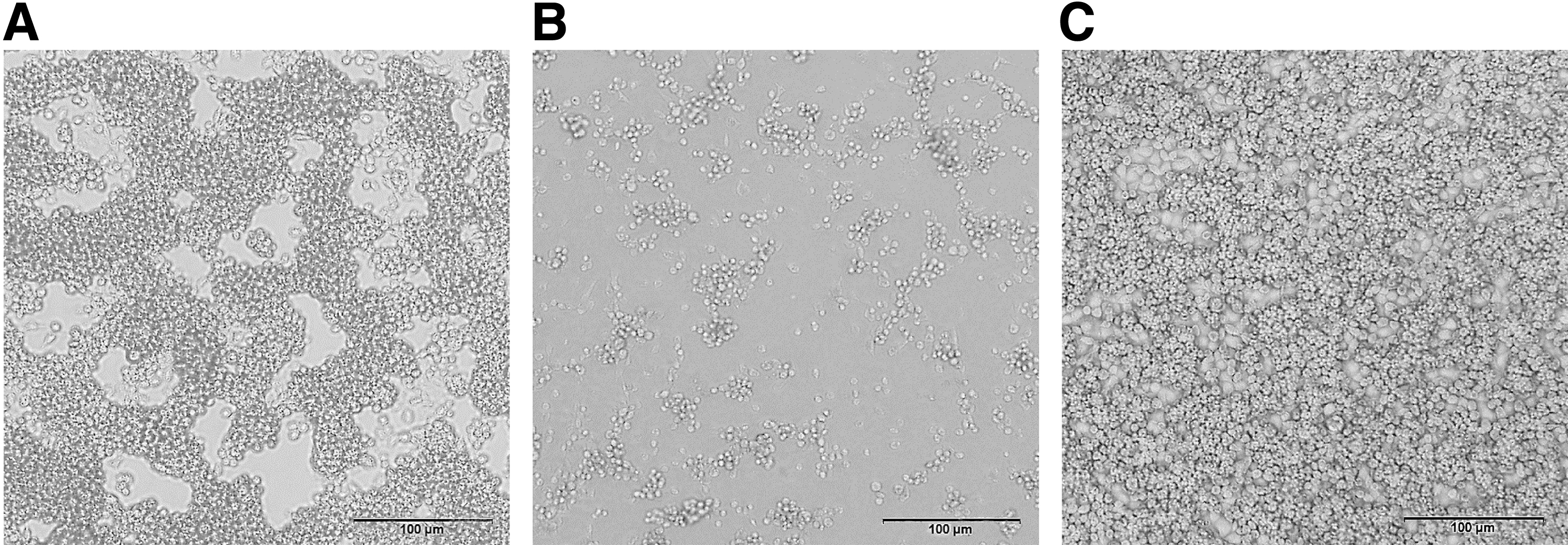
Cytopathic effect of SZ_M48 on C6/36 cells at 72 and 144 h ( × 100).
Virus identification by RT-qPCR and AGE
The RNA extracted from the supernatant of the SZ_M48 isolates tested negative for known endemic arboviruses (AKAV, BAV, BTV, EHDV, PALV, and TIBOV) in Yunnan by previously described RT-qPCR assays (Duan et al., 2022; Hofmann et al., 2008). AGE analysis demonstrated that the genome of the SZ_M48 virus consists of 12 dsRNA segments, all of which migrate separately to form a “6-5-1” pattern (Fig. 2). The RNA genome segments of KDV, BAV, LNV, BALV, and Mangshi virus in the genus Seadornavirus migrate as groups of 6-5-1, 6-6, 6-2-3-1, 7-3-2, and 6-4-2, respectively, during 1% AGE, and these patterns are thought to be characteristic for each virus species (Reuter et al., 2013; Wang et al., 2015; https://ictv.global/report/chapter/sedoreoviridae/sedoreoviridae/seadornavirus).
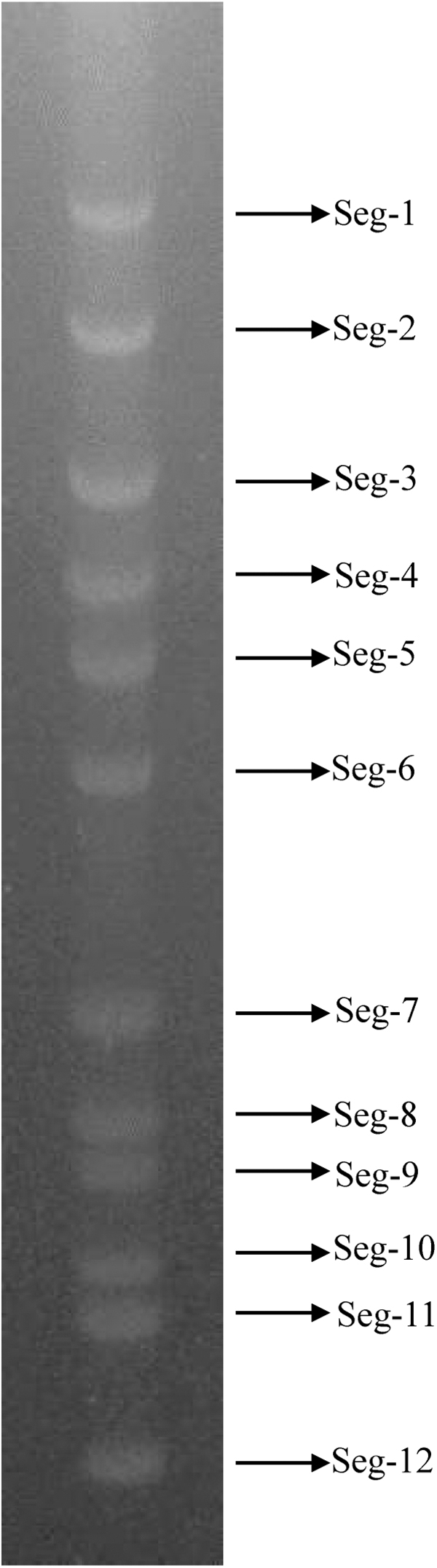
Agarose gel (1%) electrophoretic profile of the genomic dsRNAs of SZ_M48.
NGS sequencing results of SZ_M48 virus
The genomic DNA of SZ_M48 was amplified by FLAC and subjected to the HiSeq 2000 platform for NGS, resulting in 121, 619, and 750 raw reads. After quality trimming and read length filtering, the qualified reads (119, 286, 362 reads) were used for de novo sequence assembly. Nearly 98.1% of qualified reads were assembled into 12 contigs, corresponding to 12 genomic segments with an average coverage depth >8000 for each nucleotide site. All of the sequences discussed refer to the positive strand, are written in a 5′–3′ orientate on, and are submitted to the NCBI GenBank with accession numbers OR900134 to OR900145.
Genome organization and characteristics of SZ_M48 virus
The length of 1–12 segments vary from 3774 bp (Seg-1) to 757 bp (Seg-12) and their corresponding encoding proteins (1223 aa to 190 aa) are presented in Table 1. Sequence analysis of the 5′ and 3′ noncoding regions (NCRs) showed that the SZ_M48 virus shares six and three highly conserved nucleotides at the ends of the 5′- and 3′-NCRs (5′-GTAGAA and GAC-3′, respectively) in each of the 12 gene segments. Like other members of the Seadornavirus genus, the 5′ NCRs are shorter than the 3′ NCRs, only Seg-12 has a longer 5′ NCR (103 bp) than 3′ NCR (81 bp), and the first and last two nucleotides of each segment are reverse compliments.
Lengths of Double-Stranded DNA Segments 1–10, Encoded Putative Proteins, 5′ and 3′ Noncoding Regions of SZ_M48 Virus Genome
Conserved nucleotide sequences in 5′- and 3′-terminals are shown in bold.
aa, amino acid; Cap, capping enzyme; NCR, noncoding region; NS, nonstructural protein; Pol, RNA-dependent RNA polymerase; T13, outer-layer core protein; T2, inner-layer coat protein.
The 5′ and 3′ NCRs of SZ_M48 virus comprised 10.09% of the total genome, and the G + C content was 37.09%. The total genome G + C content was 44.36% and 42.55% for Mangshi virus and LNV, respectively, while it was 39.23% to 39.6% for BAV and 37.18% for KDV. (Attoui et al., 2006; Yang et al., 2023a). This clearly shows that the KDV genome has a lower G + C content than those of Mangshi virus, LNV, and BAV.
Gene sequence comparison of KDVs with complete coding sequences
The nucleotide (nt) and amino acid (aa) sequence identity analysis of all 12 segments was conducted among SZ_M48 and other 4 KDV isolates with complete coding sequences (CDSs) information (Table 2). The results show that SZ_M48 has higher levels of sequence identity with two Chinese strains (QTM27331 and SDKL1625) than with Indonesian strain (JKT-7075) and Danish strain (21164-6/M.dau/DK). The SZ_M48 has 73.9–91.2%/66.6–97.6% nt/aa identity with the JKT-7075, 82.5–92.4%/84.4–96.2% nt/aa identity with the 21164-6/M.dau/DK, 86.6–99.4%/87.4–100% nt/aa identity with the QTM27331 and 83.3–99.2%/86.0–100% nt/aa identity with the SDKL1625. Excluding the VP5 and VP8 genes, the aa identity of the remaining 10 segments of SZ_M48 are more than 90% compared with that of QTM27331 or SDKL1625.
Summary of Percentage Sequence Identities (%) of Nucleotide and Amino Acid for VP1–VP12 for SZ_M48 and Other Kadipiro Virus Respectively
These values were omitted, as the sequence of SDKL1625 strain Seg-4 (MG590149) had a stop codon in the region corresponding to another KDV ORF.
—, lack of sequences information; BAV, Banna virus; BALV, Banna-like virus; KDV, Kadipiro virus; LNV, Liao ning virus.
Phylogenetic analysis among KDVs and other members of the Seadornavirus genus
Phylogenetic relationships based on the CDSs of all segments of the SZ_M48 and the corresponding sequences from seadornaviruses (BAV, KALV, LNK, and Mangshi virus) were generated (Fig. 3). The results indicate that SZ_M48 was distinct from BAV, BALV, LNV, or Mangshi virus, and it clustered together with KDVs. SZ_M48 was most closely related to the Chinese strains in all phylogenetic trees except that in VP8 tree, it was close to the Danish strain (21164-6/M.dau/DK).
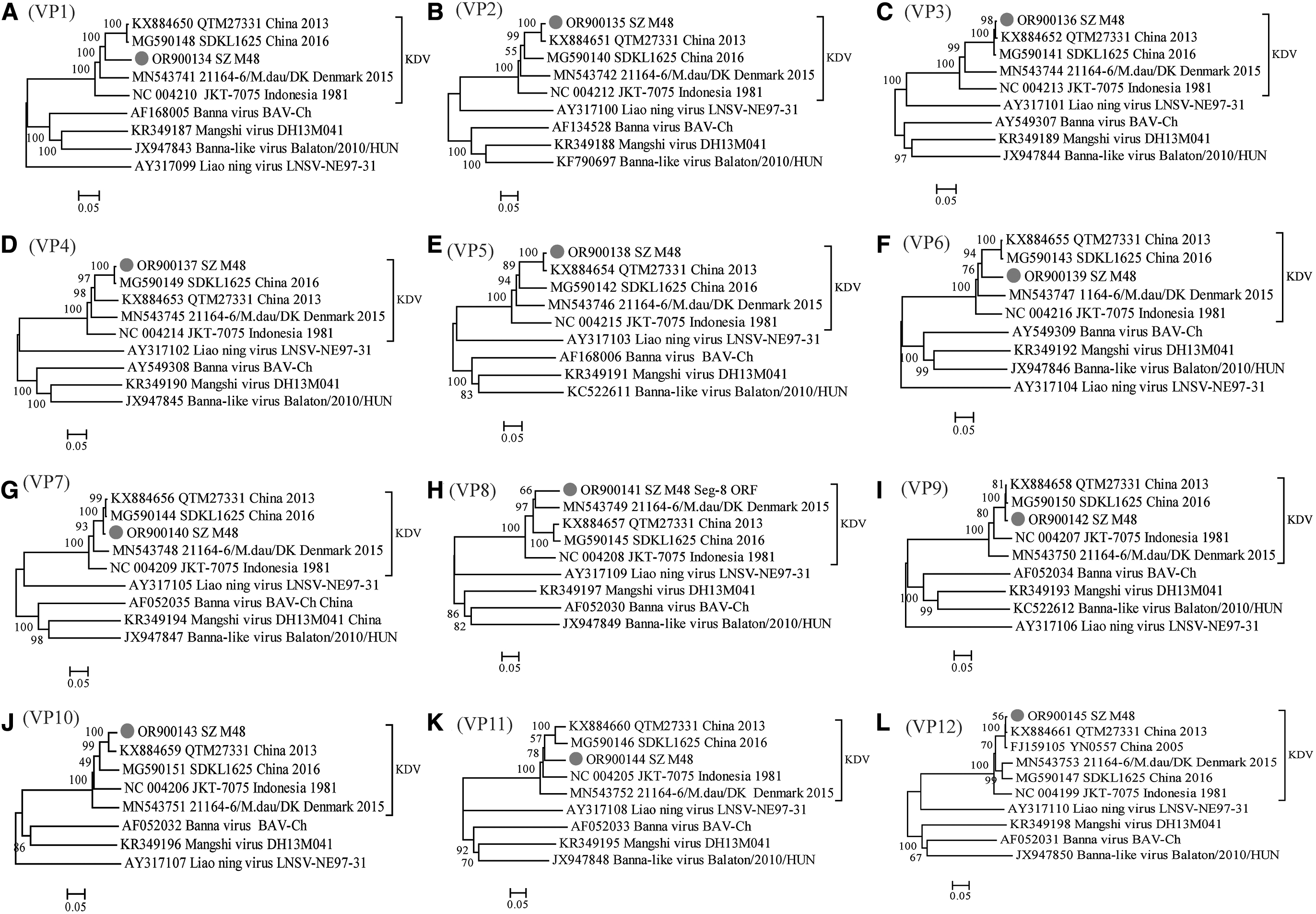
Phylogenetic analysis based on the CDSs of all segments of SZ_M48 and other members of the genus Seadornavirus. Neighbor-joining tree was constructed using p-distance determination algorithm in MEGA 6 with 1000 bootstrap replicates. Complete coding genome of each reference KDV strains was represented as “GenBank accession number_Strains number_Country (region)_year of isolation.” Outgroup virus was represented as “GenBank accession number_Virus name_Strains number.” The isolates in this study are depicted by black dots. The letters A–L stand for VP1–VP12, respectively. CDSs, complete coding sequences; KDV, Kadipiro virus.
Discussion
The genus Seadornavirus includes BAV, KDV, LNV, BALV, and Mangshi virus, of which BAV, LNV, and Mangshi virus were all first discovered in China. In this study, one strain virus (SZ_M48) was isolated from Culex tritaeniorhynchus Giles that was collected in Shizong county, southwest China in 2023, and full-genome sequencing was also performed. Some features of SZ_M48 resembled KDV, in terms of genome segment migration patterns, conserved terminal nucleotide sequences of the 5′ and 3′ NCRs, the total genome G + C content and the highest identity between their different segments, suggesting that the SZ_M48 belong to KDV.
Mosquitoes are the main vector of Seadornaviruses virus. Until now, BAV has been isolated from including 10 mosquito species in three genera (Aedes, Anopheles, and Culex), LNV has been isolated from four genera (Culex, Anopheles, Mansonia, and Aedes), Mangshi virus isolated from Culex tritaeniorhynchus Giles. (Lu et al., 2011; Prow et al., 2018; Xia et al., 2018b). KDV was first isolated from Culex fuscocephalus mosquitoes in Indonesia, and from Culex tritaeniorhynchus, Anopheles sinensis, and Armigeres subalbatus in Yunnan and Shandong provinces, China (Brown et al., 1993; Sun et al., 2009a; Zhang et al., 2018). The SZ_M48 strain in this study was also isolated from mosquitoes without blood meals. Using viral metagenomic analysis, sequencing information for strain 21164-6/M.dau/DK (Denmark) and QTM27331 (Hubei, China) was obtained from bat feces and Odonata samples, respectively. (Lazov et al., 2021; Shi et al., 2016).
Bats and Odonata will both fest on mosquitoes and mosquito larvae. We could not exclude the possibility that these two strains of KDV originated from mosquitoes that were eaten. Therefore, can we cautiously conclude that mosquitoes may be a natural vector for KDV, but further studies are needed to verify this.
KDV was first isolated from Java (latitude 6°–9° S) in Indonesia, and subsequently isolated or detected from Yunnan (21°–29° N), Hubei (29°–33° N), and Shandong (34°–38° N) provinces in China and Denmark (55°–57° N). The geographic distribution of these strains ranges from near the equator to 55°N, extending from the tropics to the north temperate zone. Therefore, the global distribution of KDV may be underestimated and larger-scale mosquito collection, pathogen testing, and serological screening are needed to determine the presence, prevalence, and distribution of KDV.
Another interesting point is why KDV can spread from low-latitude tropical zone to high-latitude temperate zone. First, increasing transnational travel, globalization, and climate warming have played an increasingly important role in the global spread of arboviruses, and these factors have also led to mosquito vectors that are now appearing in areas where they did not previously occur (Elbers et al., 2015; Hendrickx et al., 2006). In addition, the spreading route of KDV may be similar to that already reported for BAV. It has previously been reported that BAV has spread from tropical Southeast Asia to the northern temperate zone of China, possibly transported by infected mosquitoes in the prevailing winds and by the migration of infected birds (Liu et al., 2010). KDV and BAV are both members of the Seadornavirus virus, with the same vector (mosquitoes).
Therefore, the spreading route of KDV is also highly likely to be influenced by winds and bird migration. However, the spreading dynamics of KDV are not well known, and more studies are needed to confirm the spreading route of the virus and the influences in this process.
Phylogenetic analysis of KDV strains and representative strains of other members of the genus Seadornavirus was performed based on the CDSs of all segments. The phylogenetic tree shows that the KDV isolates can be divided into 3 distinct branches that appear to be geographically related. In all phylogenetic trees except these of VP8 and VP12, SZ_M48 was closely related to the Chinese strain and formed a separate branch, while the Indonesian and Danish strains each formed an independent branch. By genetic sequence comparison, it was shown that of all KDV strains, SZ_M48 has the highest sequence identity with strain QTM27331 isolated from central China's Hubei province, with 86.6–99.4%/87.4–100% nt/aa identity.
The identity between SZ_M48 and SDKL1625 isolated from Shandong Province in northern China is slightly lower than that of QTM27331, and SZ_M48 had the lowest identity with the Indonesian (JKT-7075) and Danish (21164-6/M.dau/DK) strains. Compared with Shandong in China, Indonesia, and Denmark, Yunnan is closer in latitude to Hubei. The above results revealed that the greater the geographical distance, the larger the differences in nucleic acid and amino acid sequences between KDV strains.
It has been reported that the MCC tree of LNV can be divided into 3 distinct groups that appear to be geographically related, with nucleotide and amino acid homology rates showing high identity within each group and much divergence between groups (Lu et al., 2011). Hong et al., based on the analysis of isolated BAV strains worldwide, concluded that BAVs from the same latitude regions are nearly 100% identical, whereas the differences between strains from long-distance regions with different latitudes may be large, and significantly cluster together in the phylogenetic tree according to their geographical distribution (Liu et al., 2016). Whether KDV will be similar to LAV and BAV, the genetic differences between different strains are positively correlated with geographical distribution and have evolved to produce different genotypes or topotypes to accommodate different environments or hosts?
However, it should be noted that the sequence of KDV in NCBI is limited, and hence, we can only use 5 strains for the full sequence analysis. As more KDV genomes are revealed in the future, evolutionary analysis of more strains will substantially improve the understanding of this virus.
Conclusions
This study describes the isolation and characterization of KDV strain SZ_M48, as well as its genomic analysis and phylogenetic relationship with members of the genus Seadornavirus. Sequence and phylogenetic analysis demonstrate that SZ_M48 is closely related to KDV isolates from Hunei and Shangdong in China. These discoveries have enriched our knowledge of the distribution of KDV in China, but further serological screening should be carried out to determine the presence, prevalence, and distribution of KDV.
Footnotes
Acknowledgments
We acknowledge the cooperation and enthusiasm of the Animal Disease Prevention and Control centers in Shizong county.
Authors' Contributions
Conceptualization: J.W. conceived and designed the experiments; Z.Y., Y.H., J.M., N.L., and S. L. performed the experiments; Z.Y. and Y.H. analyzed the data and wrote the article.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The background information and GenBank accession numbers of all virus strains used in this study can be found in the Supplementary Table S1.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors. Authorization for the collection of blood-sucking insect has been obtained from Institute for Yunnan Animal Science and Veterinary Institute, Kunming, China.
Author Disclosure Statement
The authors have declared that no competing interests exist.
Funding Information
This study was funded by National Natural Science Foundation of China (32260896); National Key Research and Development Program of China (2022YFC2601603); Basic Research Projects of Yunnan Province (202201AS070062); Projects funded by the central government to guide local scientific and Technological Development (202207AB110006); Open Research Fund of Chinese Academy of Sciences (2022SPCAS001). Science and Technology Talents and Platform Plan of Yunnan Province Science and Technology Department (202305AO350020).
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.