
Abstract
Background:
Over the past decade, numerous new tick-associated flavi-like viruses with segmented genomes have been discovered almost worldwide. Kindia tick virus (KITV) was first detected in Rhipicephalus geigyi ticks in West Africa in 2017. The current study aimed to detect viral RNA in tick and cattle samples collected in Guinea and to perform complete sequencing of KITV isolates and their analysis.
Methods:
Adult ticks and blood samples were collected from cattle in Coyah, Dubréka, Forécariah, and Kindia prefectures of the Republic of Guinea in 2022. These samples were tested for KITV infection by RT-PCR with primers targeting the NS5 gene. Positive probes were sequenced using Illumina technology, and their analysis was performed for obtaining complete sequences of all KITV segments.
Results:
The RNA of the KITV was detected by RT-PCR in Rh. geigyi, Rh. annulatus ticks, and blood samples of cattle. The prevalence rates for cattle were 6.6%, for Rh. annulatus 6.9%, and for Rh. geigyi ticks 10.7%. The analysis of 15 complete sequences of KITV genomes showed 99.61–99.67% identity for amino acid sequences for segments 1 and 4 and 97.88–98.83% for segments 2 and 3 with previously detected KITV isolate in Guinea in 2017. Phylogenetic analysis demonstrated that obtained KITV sequences can be classified as typical for clade A of the Jingmen tick virus (JMTV) group together with Mogiana tick virus.
Conclusion:
The KITV isolates from cattle and feeding ticks show practically full identity sequences for all four viral segments, and these sequences can be classified as clade A of the segmented flavi-like virus JMTV group.
Introduction
For the first time, a novel segmented flavi-like virus was detected in Rhipicephalus microplus ticks in the Jingmen region of China’s Hubei province, and the virus was named as the Jingmen tick virus (JMTV) in accordance with the geographical location of the discovery (Qin et al., 2014). The viruses of the JMTV group are described by the International Committee on Taxonomy of Viruses as an unclassified segmented flavi-like virus belonging to the family Flaviviridae, genus Orthoflavivirus (available from: https://ictv.global/report/). The genome of these viruses is represented by the segmented ss(+)RNA and includes four short fragments, each flanked by 5′ and 3′ untranslated regions. The total length of the genome of the segmented JMTV is approximately 11,000 nucleotides. Segments 1–3 carry one extended open reading frame (ORF) and encode NS5, VP1, and NS3 proteins, respectively. Segment 4 carries two ORFs and encodes VP2 and VP3 proteins. The NS3 and NS5 proteins have structural and functional similarities with viral proteins in nonsegmented flaviviruses. Structural proteins VP1, VP2, and VP3 do not have known homologs both among the flaviviruses and among other viruses known to date.
The Kindia tick virus (KITV) was first detected in Kindia prefecture in the Republic of Guinea in ticks in 2017 (Ternovoi et al., 2020b). Later, the KITV sequences were detected in Rh. geigyi, Rh. annulatus, and Rh. decoloratus ticks by RT-PCR, and partial fragments of the genome were sequenced with following phylogenetic analysis (Kartashov et al., 2023). Analysis of the spatial models of NS3 and NS5 of KITV also showed that these proteins had a high level of topological similarity to viral proteins of the tick-borne encephalitis and dengue viruses (Gladysheva et al., 2023). So far, KITV is the first segmented flavi-like virus described in West Africa.
In the recent decade, many novel flavi-like viruses with segmented genomes have been detected in different continents (Colmant et al., 2022). These viruses were found in Asia, Europe, South America, and Africa in a wide range of both arthropod (Amblyomma, Dermacentor, Ixodes, Rhipicephalus, Hyalomma ticks and mosquitoes) and animal hosts (bats, rodents, cattle, nonhuman primates, and humans). The JMTV-like isolates have also been detected in China and Brazil in cattle (de Souza et al., 2018). The Alongshan virus has also been found in Bos taurus in China (Wang et al., 2019a). The JMTV-like isolates were shown to circulate in East Africa, where they were found in blood of nonhuman primates in Uganda and ticks collected from cattle, sheep, and turtles in Kenya (Ladner et al., 2016; Ogola et al., 2022).
The viruses belonging to the JMTV group have been detected in blood of patients with Crimean–Congo hemorrhagic fever in Kosovo and Russia (Emmerich et al., 2018; Ternovoi et al., 2020a). The JMTV reproduces in human skin biopsy specimens after a tick bite; eight serologically positive patients having severe clinical symptoms and a history of tick bites were detected in a retrospective study (Jia et al., 2019). Anti-JMTV antibodies were also detected in 18–37% of population in several regions of China (Qin et al., 2014; Shi et al., 2021). The Alongshan virus was found to be associated with human febrile illness in northeastern China (Wang et al., 2019b). Hence, the pathogenicity of some segmented flavi-like viruses for farm animals and humans has been demonstrated. However, this information is fragmentary and limited.
The study objective was to search for a molecular genetic marker of KITV in ixodid ticks collected from cattle, evaluate the presence of KITV RNA in blood of cattle, as well as perform complete sequencing of the KITV genomes from tick and cattle samples and their phylogenetic analysis.
Materials and Methods
Study sites, tick sampling, and morphological identification
Adult ticks and animal blood samples were collected from cattle in Coyah, Dubréka, Forécariah, Kindia prefectures, and Telimele province of Kindia in the Republic of Guinea in 2022 (Fig. 1). Following surface sterilization with 70% ethanol and washing with deionized water to remove foreign particles from animal skin, the collected ticks were identified morphologically to species (Walker et al., 2014). Ticks were mechanically homogenized in 1.5 mL microcentrifuge tubes with zirconia beads (0.5 mm in diameter) for 1.5 min in a Bioprep-24 homogenizer (Hangzhou Allsheng, China). Animal blood samples were collected into EDTA tubes and also subjected to RT-PCR testing.
The map of the Republic of Guinea presenting sample collection sites in Coyah, Dubréka, Forécariah, and Kindia prefectures. The collection sites are marked with asterisks.
RNA extraction and PCR screening
Total RNA was extracted from 150 µL of tick homogenates (homogenization described above) or 300 µL of blood serum from cattle using the ExtractRNA reagent/TRIzol analog (Evrogen, Russia) following the manufacturer’s instructions. Reverse transcription was carried out for 5 µL of the isolated RNA using the Mint-2 cDNA synthesis kit (Evrogen). Samples were screened for KITV infection by PCR using primers targeting the NS5 gene (Kartashov et al., 2023).
Library preparation and next-generation sequencing
Viral RNA was extracted from KITV-positive samples using a QIAamp Viral RNA Mini Kit (Qiagen, Germany). Multiprimer target PCR with specific primers and high-precision Phusion DNA polymerase (Thermo FS, USA) was used to enrich the fragment library of the KITV genome. Purification of the PCR products was carried out using GeneJET Gel Extraction and DNA Cleanup Micro Kit (Thermo FS). Total RNA/cDNA was quantified with a Qubit RNA Assay Kit (Invitrogen Co., USA) following the manufacturer’s instructions. Sequencing was performed using a MiSeq Reagent Kit v3 for 600 cycles. Cutadapt (version 48 1.18) and SAMtools (version 0.1.18) were used to remove the Illumina adaptors and duplicate reads. The contigs were assembled de novo using the MIRA assembler with default parameters (version 4.9.6). High-throughput sequencing data were processed using the BLAST-based taxonomic read identification algorithm (McGinnis and Madden, 2004).
Phylogenetic analysis
Maximum likelihood (ML) phylogenetic trees were estimated using PhyML version 2.2.4 with the best-fit model determined by Modeltest implemented in MEGA-X version 10.2.5 with nodal support being assessed through 1000 bootstrap replications.
Statistical analysis
For estimating KITV prevalence in individual tick species, a frequentist model was used to implement ML analysis in an online platform, Epi-Tools epidemiological calculator (available from: http://epitools.ausvet.com.au/).
Sequence accession numbers
KITV coding complete genome sequences were deposited in GenBank under the accession numbers OP612399-OP612413 for segment 1; OP612414-OP612428 for segment 2; OP612429-OP612443 for segment 3; and OP612444-OP612458 for segment 4.
Results
The study analyzed 45 blood samples from free-ranging adult cattle (B. taurus) in the Coyah, Dubréka, Forécariah, Kindia prefectures, and Telimele province of Kindia, as well as 156 individual samples of ticks (Amblyomma variegatum, Rh. geigyi, Rh. annulatus, Rh. decoloratus) collected from cattle (Supplementary Table S1). The KITV RNA was detected in three blood samples from cattle and in 12 Rh. geigyi and Rh. annulatus ticks. The prevalence rate for cattle was 6.6%, for Rh. annulatus 6.9%, and for Rh. geigyi ticks 10.7% (Table 1).
RT
CI, confidence interval; KITV, Kindia tick virus.
The analysis of all four segment sequences for 15 KITV isolates showed a high level of identity: 96.31–99.03% for nucleotide and 97.88–99.67% for a.a. sequences with the KITV/2017/1 isolate (Supplementary Table S2). The a.a. sequences of the recent KITV isolated in 2022 from cattle blood and ticks taken from these cattle show complete identity for segment 1 (NS5-like) and the VP2 polypeptide encoded by segment 4 (Supplementary Table S3). In segment 2 encoding VP1, the isolates had a three single a.a. substitutions between each other in position 363, 523, and 728. These three substitutions are similar to KITV/2017/1 isolate. In contrast, when comparing the KITV/2017/1 isolate with modern KITV isolates, we found 11–16 a.a. substitutions in segment 2 (VP1) and 3 (NS3-like), respectively.
The results of phylogenetic analysis obtained KITV sequences for each segment are presented in Figure 2. All obtained KITV sequences are closely related to the Mogiana tick virus (MGTV) and they can be classified as typical for clade A of the JMTV group.
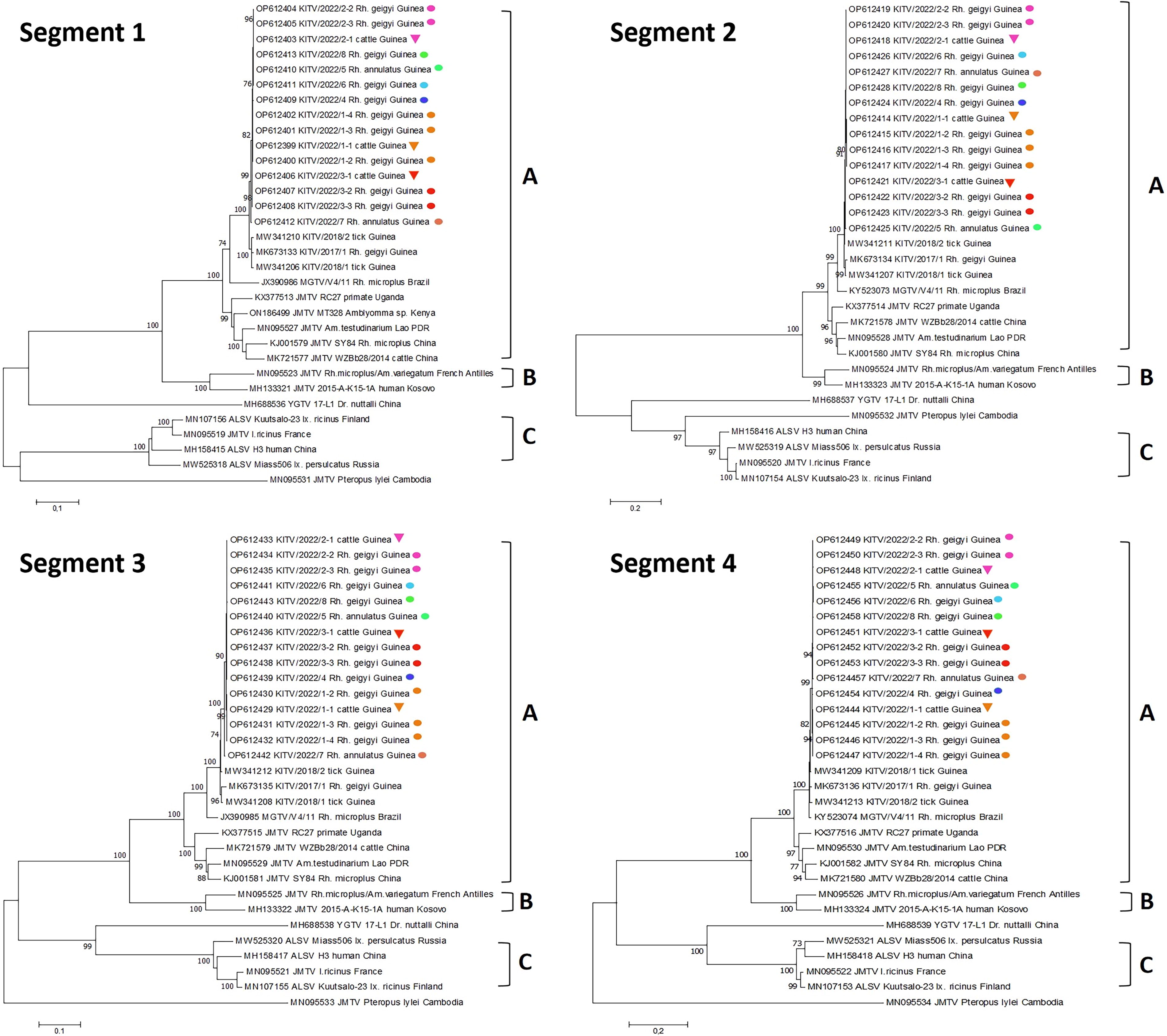
Phylogenetic trees constructed for four segments of the KITV. Triangles represent KITV sequences obtained from cattle blood. Circles represent sequences obtained from ticks. Sequences from the cow and the ticks removed from that cow are colored the same. KITV, Kindia tick virus.
Discussion
The genome of JMTV has four segments (Qin et al., 2014). The segment 1 and 3 encoded flavi-like NS3 and NS5 polypeptides, which in all likelihood could have arisen during the evolution of flavivirus genomes. The two unique segments that encoded structural proteins VP1, VP2, and VP3 (segments 2 and 4) were acquired independently of an as-yet unidentified ancestor. Thus, the hypothetical existence of two ancestors for this group and their emergence through reassortment of genomes is possible. Nevertheless, the nucleotide sequences of four segments demonstrate that these segmented genomes are similar for geographically different JMTV isolates circulating in Europe, Asia, Africa, and South America (Wu et al., 2023).
All the obtained KITV sequences formed a monophyletic clade closely related to the MGTV isolated from Rh. microplus ticks collected from cattle in Brazil (Villa et al., 2017; Pascoal et al., 2019). Flavi-like segmented viruses are currently divided into at least three clades (Colmant et al., 2022). The KITV virus is classified as belonging to clade A, which also includes the MGTV (Brazil), JMTV (China and Asia), and JMTV (Uganda) variants found in nonhuman primates in East Africa (Fig. 2). Phylogenetic analysis suggests that clade A can be divided into subclade A1 and A2. Subclade A1 includes KITV and MGTV isolates, which have been found in ticks and cows on the Atlantic coast of Africa and South America. Clade B includes JMTV isolates from the Antilles and Kosovo variants of JMTV (MH133313–MH133316) isolated from Congo–Crimean hemorrhagic fever patients. Clade C combines variants of the Alongshan virus found in domestic animals and humans in China and later detected in Europe and Russia.
Available data indicate the possible introduction of the KITV ancestors to the American continent from the western regions of Africa and/or Europe with livestock. The cattle have been introduced to this continent relatively recently, thus suggesting that the common ancestor for these viruses was exported along with cows about 500 years ago to the American continent across the Atlantic Ocean (McTavish et al., 2013). Furthermore, the KITV and MGTV have been spreading in nature of two continents independently of each other. Their minor genetic differences are accumulated over a relatively short time and are probably related to human agricultural activity.
The discovery of various genetic variants of JMTV from clades B and A2 in Africa (Guinea, Kenya, and Uganda) and Asia (China) suggests that JMTV ancestors may exist in the southern regions of these continents. It may be possible that after the end of the ice age, JMTV spread widely across the natural biotopes of northern Eurasia. It is likely that ticks and pets could have provided such a wide spread of JMTV-like viruses. However, to better understand this JMTV widespread in global size, one needs to investigate the potential role of bats, rodents, and especially migratory birds.
It is interesting to note that whole genome sequencing of KITV showed practically complete identity of the viral genomes in ticks and cows. This suggests that cattle may be involved in spreading of the KITV. Previously, JMTVs have also been detected in cows and water bulls in Southeast Asia (Colmant et al., 2022). The Alongshan virus has also been detected in sheep and cattle in China (Wang et al., 2019a). Detection of segmented viruses in blood of domestic animals suggests possible human infection through contact with animals, their blood, milk, and meat products.
Conclusions
KITV is the first segmented flavi-like virus found in West Africa. The genetic material of KITV was detected by RT-PCR in Rh. geigyi and Rh. annulatus ticks collected from cattle, as well as in the blood of cows in 2022. The infection rate was 6.9–10.7%. Fifteen KITV isolates from cattle blood (3 isolates) and feeding ticks (12 isolates) were sequenced. The KITV complete genome (four segments) showed a high level of their identity with previously described KITV in Guinea in 2017. Phylogenetic analysis also demonstrated that studied KITV isolates can be classified as typical for clade A, where they together MGTV are possible forming separate genogroup. Thus, the obtained results indicate the need for additional studies aimed at understanding on the molecular epidemiology of KITV in West Africa.
Ethical Approval and Consent to Participate
This article does not describe any studies involving humans as a test subject. The study protocol was approved by the decision of the National Ethics Committee of the Ministry of Health of the Republic of Guinea (Protocol No. 129/CNERS/16 of August 31, 2015).
Footnotes
Acknowledgments
The authors would like to gratefully acknowledge support from the Russian Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing.
Authors’ Contributions
M.Y.K.: Conceptualization (lead), writing—original draft (equal), and validation. E.I.K., E.V.N., and K.S.Z.: Collection and analysis of samples (equal), methodology, and investigation. A.N.S.: Sequence analysis and software (lead). S.B.: Collection and analysis samples (equal), formal analysis, resources, and supervision. V.A.T.: Methodology, formal analysis, resources, and supervision (equal). V.B.L.: Conceptualization and writing—review and editing (equal).
Author Disclosure Statement
The authors have no conflicts of interest associated with this study.
Funding Information
This research was conducted within the framework of the Federal project Sanitary Shield - Safety for Health (Prevention, Detection, Responce).
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.