
Abstract
Background:
Zika virus (ZIKV) belongs to the Flavivirus genus of the Flaviviridae family. It is an enveloped RNA virus that enters host cells via receptor-mediated endocytosis. The interactions between viral proteins and particular receptors on the host cell surface is the initial step of the virus life cycle, which represents the key targets for antiviral therapeutic.
Materials and Methods:
This review highlights a variety of cell types infected by ZIKV, including human radial glial cells, endothelial cells, neural progenitor cells, astrocytes, microglia, and Sertoli cells. The cellular molecules involved in the entry process of ZIKV are detailed, and the advances in the development of chemical compounds and neutralizing antibodies targeting the ZIKV entry process are described.
Results:
The interactions of ZIKV with cellular molecules in various host cells during virus entry are reviewed, as the targets of the development of antiviral therapeutics.
Conclusion:
The entry of ZIKV into host cells involves complicated mechanisms, which remain to be further explored to facilitate the development of antiviral reagents.
Introduction
Zika virus (ZIKV) is a human-infecting, single-stranded positive RNA flavivirus transmitted by mosquitoes. ZIKV was first isolated in 1947 from the serum of a sentinel Rhesus monkey in Uganda’s Zika woodland (Dick et al., 1952). ZIKV gained significant attention almost 60 years later due to major outbreaks in Micronesia, the South Pacific Islands, and South America (Duffy et al., 2009; Zanluca et al., 2015). A mosquito vector predominantly spreads the virus, typically Aedes spp., although it can also be transmitted through sexual contact or via blood transfusion (Musso et al., 2014). The outbreak of ZIKV-induced fetal microcephaly and congenital Zika syndrome has prompted extensive research into its cell tropism in order to formulate effective antiviral therapeutics (Garcez et al., 2016; Mlakar et al., 2016).
Over the past 50 years, ZIKV strains have been sporadically isolated from Africa, Asia, and the America (Berthet et al., 2014; Ladner et al., 2016). The genomic RNA of the ZIKV strain is about 10,807 nucleotides (nt) long. It consists of a 10,272-nt open reading frame (ORF) flanked by a 107-nt 5′ noncoding region (NCR) and a 428-nt 3′ NCR. The ORF encodes a 3,423-amino acid (aa) polyprotein that is predicted to be cleaved into three structural proteins (capsid protein, prM protein, envelope protein) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5 protein) (Yun et al., 2016). The viral genome is encapsulated in the capsid (C) protein, followed by the Membrane (M) protein and the outermost layer of Envelope (Env) glycoprotein (Sirohi et al., 2016).
Flaviviruses enter host cells through receptor-mediated endocytosis and then move into endosomes. In the acidic environment of the endosomes, major conformational changes occur in the Env protein of the virus, inducing fusion of the viral and host cell membranes (Modis et al., 2004). The phosphatidylserine receptors T cell immunoglobulin mucin domain (TIM) and TAM (Tyro3, Axl, and Mer) are among the primary candidate cellular receptors (Davis et al., 2006; Meertens et al., 2012). ZIKV exploits Axl and Tyro3 receptors, which are ubiquitously expressed on various cell types, such as human radial glial cells, endothelial cells (ECs), NPCs, astrocytes, microglia, Sertoli cells (SC), and Hofbauer cells, for internalization (Hamel et al., 2015; Papa et al., 2017; Strange et al., 2019). The cell surface receptor α-2,3-linked sialic acid has also been reported to play a role in the internalization of ZIKV into human-induced pluripotent stem cells (hiPSCs)-derived neural progenitor cells (NPCs) (Tan et al., 2019). Also, glycosaminoglycans (GAGs) and lectins are considered the most important cellular factors for the attachment of flaviviruses (Kim et al., 2017). Because ZIKV has received far less attention than other emerging arboviruses, such as yellow fever, dengue (DENV), West Nile (WNV), Japanese encephalitis, and Chikungunya viruses, the pathogenesis of ZIKV infection remains poorly understood. ZIKV infection has become a serious public health issue, and there is no effective medicine or vaccine licensed for treating or preventing ZIKV infection. So, this review focuses on recent progress in understanding ZIKV attachment to cells, internalization, and membrane fusion, with specific emphasis on cellular receptors, which represent the key targets for antiviral development.
The Role of Axl in ZIKV Infection of a Variety of Cells
Flaviviruses usually enter the host cell via Env-mediated endocytosis (van der Schaar et al., 2008). Many cellular molecules are involved in the process of ZIKV entrance into different host cell types, such as phosphatidylserine (PS) receptors, T cell immunoglobulin and mucin domain 1 (TIM1), and the TAM receptors (Tyro3 and Axl), or dendritic cell-specific intercellular adhesion molecule-3 grabbing nonintegrin (DC-SIGN) (Tabata et al., 2016).
TAM receptor Axl and its ligand Gas6 are important in ZIKV infection of glial cells such as microglia and astrocytes. During ZIKV infection of brain cells, Axl/Gas6 performs a dual function: Axl indirectly binds ZIKV via Gas6 bridging and mediates ZIKV endocytosis via the clathrin-mediated endocytosis (CME) route. Axl is a signaling molecule activated by ZIKV-Gas6 complexes during viral entry to dampen innate immunity (Meertens et al., 2017). The vesicles containing ZIKV are then sent to Rab5+ endosomes. The ZIKV/Gas6 complex activates Axl kinase activity to downmodulate interferon (IFN) signaling and facilitates viral infection (Meertens et al., 2017) (Fig. 1). Axl negatively regulates IFN signaling by inhibiting the expression of IFN-stimulated genes (ISGs), which are crucial for antiviral responses. Axl can limit the production of type I IFNs through a negative feedback loop, reducing the host’s ability to mount an effective antiviral response. Additionally, Axl can modulate the IFN signaling pathway by downregulating the activation of IFN signaling genes, including type I IFNs and IFN-stimulating genes. This modulation of IFN signaling by Axl can impact the host’s ability to combat viral infections effectively (Zhang et al., 2022).

ZIKV entry receptors. ZIKV interacts with Gas6 and enters cells through Axl. Following that, the virus suppresses innate immunity by activating Axl kinase, which downmodulates interferon signaling and facilitates infection (Meertens et al., 2017) or promotes the transcription of TLR3, RIG-I, and MDA5, as well as many interferon-stimulated genes (ISGs) in human skin fibroblasts (Hamel et al., 2015) and the formation of autophagosomes, which was associated with enhanced virus replication. TLR3, Toll-like receptor 3; ZIKV, Zika virus.
ZIKV infection induced the transcription of Toll-like receptor 3 (TLR3), RIG-I, and MDA5. It also resulted in the upregulation of ISGs, including OAS2, ISG15, and MX1, which ultimately caused a notable increase in the expression of the beta IFN gene (Hamel et al., 2015) (Fig. 1). The effects of Axl on ZIKV infection differ between glial and skin cells. Axl promotes viral entry in glial cells and modulates immune responses, facilitating infection (Meertens et al., 2017). Conversely, in skin cells, Axl is an attachment factor for ZIKV on the cell surface, requiring endocytosis and acidic pH for productive infection (Hamel et al., 2015; Persaud et al., 2018). Axl also promotes the formation of autophagosomes in skin cells, which is associated with enhanced virus replication (Hamel et al., 2015). The distinct roles of Axl in glial versus skin cells may be attributed to cell-specific factors influencing viral entry mechanisms and immune responses.
Axl expressed in glial cells in the developing brain mediates ZIKV infection
Axl protein is expressed in developing human brain cells such as radial glial, astrocytes, ECs, and microglia, making them more susceptible to ZIKV infection (Nowakowski et al., 2016). Axl is expressed in an extremely reproducible pattern throughout the cortex at mid-neurogenesis, with strong expression bordering the lateral ventricle and the outer subventricular zone (OSVZ). Furthermore, prominent Axl immunostaining defined brain capillaries, compatible with Axl expression in ECs identified by single-cell analysis. Axl expression persisted in radial glia throughout neurogenesis, in capillaries and astrocytes until term but was largely missing in SATB2-expressing neurons, even at later stages of development (Nowakowski et al., 2016).
ZIKV infectivity in the developing human brain was explored using organotypic cultures of primary human tissue (Retallack et al., 2016). Human cortical tissue slices were cultured for 72 h and infected with three ZIKV strains from different outbreaks (Cambodia 2010 [ZIKV-CAM], Brazil 2015 [ZIKV-BR], and Puerto Rico 2015 [ZIKV-PR]), and the infectivity was detected by immunostaining for the flavivirus envelope protein (Env). Substantial infection frequencies were found in the ventricular and subventricular zones in mid-neurogenesis samples. Both ventricular and outer radial glial cells were preferentially infected. U87 glioblastoma cell expresses astrocyte marker genes, including Axl, at high levels, and is easily infected by ZIKV with high viral production at 48 h post-infection (hpi) and a strong cytopathic impact at 72 hpi. Knock-out of the Axl gene in U87 significantly decreased the infection of ZIKV, indicating the relevance of AXL with ZIKV infection in this cell type (Retallack et al., 2016).
Endothelial cells
ZIKV-infected IFN alpha receptor-knockout (Ifnar −/− ) pregnant mice (mice lacking type I IFN signaling) have signs of vascular damage in the placenta, such as abnormal shape, destruction of the placental microvasculature, and fewer fetal blood vessels, indicating in vivo ZIKV infection of fetal ECs (Miner et al., 2016).
The infectivity of ZIKV was tested using cultures of ECs isolated from human umbilical veins (HUVECs), aorta (HAoECs), coronary artery (HCoECs), saphenous vein (HSaVECs), and lymphatic endothelial cells (Liu et al., 2016). Both African (AF) and South American (SA) strains of ZIKV infected all vascular ECs from numerous donors, with HUVECs being much more susceptible. A polyclonal antihuman Axl antibody inhibits ZIKV infection in human cerebral microvascular endothelial cells (hCMEC/D3) and HUVECs while not impacting viral binding to cells (Liu et al., 2016). Also, in human embryonic kidney 293 (HEK293T) cells, the absence of Axl or diminished Axl kinase activity does not affect virus binding to the cells but significantly reduces viral infection, and loss of Axl kinase activity partially reduces viral infection. These findings imply that Axl is not essential for virus attachment to cells, but is required for functional virus infection of cells, which is partially associated with the enzymatic activity of Axl (Liu et al., 2016).
Astrocytes
Astrocytes are among the first brain cell types to be attacked by ZIKV after peripheral infection of newborn immunocompetent mice (van den Pol et al., 2017). Astrocytes mount a fast IFN response, limiting viral propagation (Lindqvist et al., 2016). Glial fibrillary acidic protein staining revealed that ZIKV infection caused a substantial rise in the number of astrocytes in mice brains at postnatal day 3 (P3), indicating astrogliosis and brain injury (Shao et al., 2016). Axl is a key receptor for ZIKV entry into astrocytes, which is mediated by the binding of the Axl ligand growth arrest-specific 6 (Gas6) (Meertens et al., 2017) (Fig. 1).
Axl interference with type I IFN signaling in astrocytes. In Axl −/− human astrocytoma cell line (U-251MG), IFN and ISG production were rapidly up-regulated in response to ZIKV infection (Chen et al., 2018). Genetic ablation of Axl may generate a faster response to ZIKV infection. This rapid response of type I IFN signaling, rather than its intrinsic expression level, proved crucial in avoiding productive ZIKV infection. These results supported that Axl promotes ZIKV infection by antagonizing type I IFN signaling (Chen et al., 2018).
Human fetal astrocytes (HFAs) can support chronic ZIKV infection with persistent viral shedding for at least one month (Limonta et al., 2018). HFAs had lower cell surface expression of Axl than human lung cancer cells (A549). The specific Axl inhibitor R428, the peptide duramycin, and the Axl antibody decreased ZIKV infection of HFAs, demonstrating the relevance of this protein in viral entry and replication in HFAs. In addition, IFN response also plays a crucial role in ZIKV infection of the HFAs. A substantial decline in ZIKV reproduction and titer levels was detected among HFAs receiving pretreatment with both IFNs (IFN-α and IFN-γ) (Limonta et al., 2018). The stochastic nature of cellular antiviral response may underlie the ability of ZIKV to spread in HFAs cultures despite a sustained antiviral response. Once the virus infects, it can use efficient countermeasures against the cellular antiviral defense (IFN response), thus enabling it to replicate (Kumar et al., 2016) productively.
Neural progenitor cells
NPCs, an essential population of the developing embryonic brain, have been identified as a direct target of ZIKV (Shao et al., 2016). ZIKV can efficiently invade human embryonic cortical neural precursor cells (Tang et al., 2016). Other cells, such as human embryonic stem cells and hiPSCs were also infected. ZIKV infection caused considerably higher caspase-3 activation in hNPCs, indicating increased cell death. ZIKV infection of hNPCs causes inhibited proliferation of this cell type (Tang et al., 2016).
The primary cause of postnatal growth restriction, including microcephaly, is thought to be disruption of NPC differentiation, where ZIKV infection affects NPC proliferation and differentiation as shown in a mouse model (Li et al., 2016). ZIKV infection significantly reduced the number of mitotic cells in the mice brains’ ventricular zone (VZ), which was accompanied by more centrosomes facing away from the nuclei. ZIKV infection suppresses NPC proliferation, the transition of Pax6+ radial glial cells to Tbr2+ interneuron precursors (IPCs), and NPC differentiation (Li et al., 2016), which are in agreement with the finding that ZIKV infection leads to S phase arrest of hiPSCs (Tang et al., 2016) and that proliferating NPCs exhibit a much longer S phase than those committed to neurogenesis (Arai et al., 2011).
Neurodevelopmental RNA-binding protein Musashi-1 (MSI1) was significantly expressed in neural progenitors in the embryonic brains of human fetuses but not in mature neurons (Chavali et al., 2017). MSI1 gene has also been shown to be mutated in people with recessive primary microcephaly (Chavali et al., 2017). MSI1 in vitro can boost UTR-driven translation of the ZIKV genome, showing that MSI1 plays an important role in both ZIKV infection and the development of fetal microcephaly (Chavali et al., 2017). Surprisingly, removing Axl via genome editing in hiPSC-derived NPCs and the resultant cerebral organoids did not affect ZIKV infection and pathology, implying that Axl is not required for ZIKV infection in human NPCs (Wells et al., 2016). Hence, there could be additional unknown receptors that aid ZIKV entry into specific cells.
Microglial cells
Microglial cells are very susceptible to ZIKV infection. The ability of microglia cells to produce ZIKV progeny in vitro was confirmed using ZIKV-infected cell line CHME-5 (Diop et al., 2018). ZIKV-specific RNA was also found in ZIKV-infected mice microglia (Wang et al., 2018). ZIKV infection causes inflammatory responses in microglia by producing TNF-α, IL-6, IL-1β, and inducible nitric oxide synthase (iNOS) (Wang et al., 2018). According to single-cell mRNA sequencing analysis, the ZIKV entry receptor Axl is enriched in microglia, radial glia, and astrocytes in the developing human cortex (Nowakowski et al., 2016).
Axl-mediated ZIKV infection is dependent on Gas6 (Meertens et al., 2017). MYD1 is an engineered Axl decoy receptor with a high affinity for human Gas6 and blocks the ligand-receptor interaction by completely neutralizing Gas6 (Kariolis et al., 2014). Also, MYD1 inhibits ZIKV infection by sequestering Gas6 and preventing virus binding to Axl. Gas6 is the key driver for ZIKV infection in glial cells. The anti-Gas6 polyclonal antibody suppressed ZIKV infection. Overall, these outcomes imply that the decoy Axl receptor MYD1 prevents ZIKV infection by binding to Gas6 and blocking the virus from attaching to Axl (Meertens et al., 2017).
Human Sertoli cells
Animal models have revealed ZIKV to be persistent in testicular tissues for up to 4 weeks (Govero et al., 2016). ZIKV infection causes severe testicular inflammation, atrophy, and sterility in male mice (Uraki et al., 2017). The impact of ZIKV infection on major cell types in the human testes was examined by measuring virus replication in human SCs and Leydig cells (LC) (Kumar et al., 2018). SCs allowed both African and American strains of ZIKV to replicate at high levels, indicating that SCs were substantially more permissive to ZIKV (Kumar et al., 2018).
During ZIKV infection, activation of innate immune genes, particularly IFN-λ2 and oligoadenylate synthase 2 (OAS2), was significantly lower in SCs, implying that the elevated vulnerability of these cells to ZIKV is most likely owing to a suppressed antiviral response (Kumar et al., 2018). While anti-Axl antibodies dramatically reduced ZIKV infection, antibodies targeting other receptors (TIM1, TIM4, Mer, and Tyro3) had no significant effect on ZIKV replication (Kumar et al., 2018). In SCs, the Axl inhibitor R428 and the phosphatidyl serine/phosphatidyl ethanolamine (PS/PE)-binding compound duramycin repressed ZIKV replication in a dose-dependent manner, demonstrating this receptor’s significance in ZIKV infection of SCs (Kumar et al., 2018).
The infectivity of ZIKV in various testicular cell types was also compared, including primary human SC, LC, mixed seminiferous tubule cells containing SC, spermatogonial stem cells (SSC), and peritubular myoid cells (PMC) (Strange et al., 2019). The immunofluorescence assay proved that SC was infected with ZIKV, whereas LC showed low immunoreactivity to ZIKV antigen. SSC and PMC were immunoreactive to the ZIKV-E antigen (Strange et al., 2019). LC has significantly lower levels of Axl protein. These outcomes suggested multiple cell types in the seminiferous tubule compartment can support infection, In contrast, LC, an important cell type in the interstitial space, is resistant to ZIKV infection, and differences in Axl protein expression may explain some of the differences in ZIKV infectivity between SC and LC (Strange et al., 2019).
Mice model infected by ZIKV
Axl receptor is not the virus’s primary entrance receptor. The treatment with an IFN-α-receptor (IFNAR)-blocking antibody makes pregnant C57BL/6 mice more susceptible to ZIKV infection (Hastings et al., 2017), and the fetuses have a lower intrauterine growth rate than control mice (Miner et al., 2016). This pregnancy model was used to see if Axl or another TAM receptor, Mertk, participated in ZIKV transmission or replication in fetal organs (Hastings et al., 2017). Pregnant WT, Axl−/−, Mertk−/−, and Axl−/−Mertk−/− dams were treated with IFNAR-blocking antibodies and subcutaneously inoculated with a Brazilian strain (Paraiba 2015) of ZIKV. In pregnant dams, ZIKV RNA levels in the brains and spleens of WT, Axl−/−, and Mertk−/− mice had no difference; however, Axl−/− dams had a substantial rise in serum ZIKV titer. Similarly, ZIKV RNA levels in the brains of IFNAR-blocking antibody-treated nonpregnant female Axl−/− mice were comparable to WT mice (Hastings et al., 2017).
Also, situ hybridization with ZIKV-specific RNA probes was performed to explore if TAM receptors altered ZIKV infection tropism in the placenta (Hastings et al., 2017). The placenta is divided into two layers: the junctional zone, made up of spongiotrophoblasts and invasive glycogen cells, and the labyrinth zone, composed of cytotrophoblasts and syncytial trophoblasts, as fetal-derived blood vessels (Coan et al., 2005). Situ hybridization for ZIKV RNA exhibited similar staining patterns, with scattered positive cells in the junctional zone and a lack of substantial positivity in the labyrinth zone; comparable numbers of positive cells were observed in placentas taken from WT, Axl−/−, and Axl−/− Mertk−/− mice. These outcomes showed that Axl and Mertk are not required for ZIKV infection in mice treated with IFNAR-blocking antibodies. Furthermore, virus transplacental transmission and propagation in the placenta and fetus can occur regardless of Axl or Mertk receptors (Hastings et al., 2017).
Other Cellular Factors Involved in ZIKV Entry
Neural cell adhesion molecule
Neural cell adhesion molecule (NCAM1) was shown to be involved in the ZIKV entry. NCAM1 is highly expressed in the brain. Overexpression of NCAM1 in HEK293T cells boosted viral attachment and entrance (Fig. 2), but deletion of NCAM1 in glioblastoma cells (U-251 MG) dramatically lowered ZIKV infection (Srivastava et al., 2020). NCAM1 can interact with ZIKV Env in a coimmunoprecipitation assay, in which heat shock protein family A member 8 (HSPA8) is also involved. Depletion of HSPA8 in U-251 MG cells using CRISPR/Cas9 technique remarkably attenuated ZIKV infection. Furthermore, NCAM1 extracellular domain (ECD) protein and anti-NCAM1 antibody were employed to compete and inhibit NCAM1 binding activity, respectively. Preincubation with both reagents significantly inhibited ZIKV binding and entrance into U-251 MG cells (Srivastava et al., 2020).
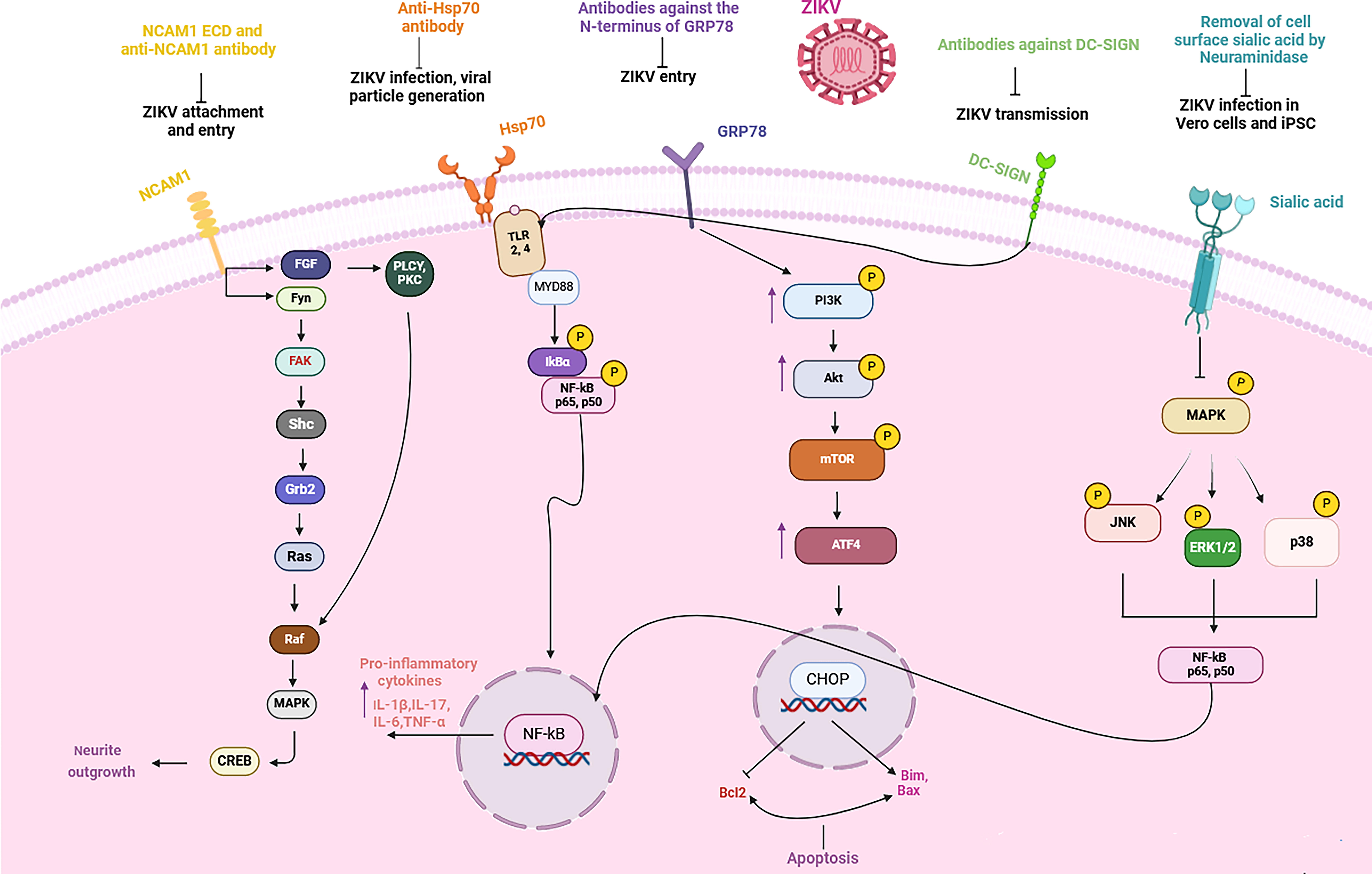
Cellular factors involved in ZIKV entry. NCAM1 is localized to the plasma membrane and cytosol (Wobst et al., 2015). The major function of NCAM1 is to regulate cell-cell adhesion, neurite outgrowth (Ditlevsen and Kolkova, 2010; Kleene et al., 2010), synaptic plasticity (Venero et al., 2006), and learning and memory in the nervous system (Vukojevic et al., 2020). Hsp70 is localized in the cytoplasm, nucleus (Arispe et al., 2002), mitochondrial matrix, endoplasmic reticulum (Ngosuwan et al., 2003), and in the plasma membrane (Vega et al., 2008), where they are involved in protein metabolism and translocation (Mouawad et al., 2023). Hsp70 acts as a molecular chaperone involved in protein folding, refolding of misfolded proteins, prevention of protein aggregation, and protein transport across membranes. The major signal pathway utilized by extracellular Hsp70 is the MyD88/IRAK/NF-κB pathway, which is typically activated by Toll-like receptors (TLRs) (Asea et al., 2002; Hulina et al., 2018). Glucose-regulating protein 78 (GRP78) is a molecular chaperone protein that translocates to the cytosol, nucleus, mitochondria, plasma membrane, and extracellular space (Sun et al., 2006; Vig et al., 2019). Also, GRP78 acts as a chaperone protein, assisting in the folding and assembly of proteins in the ER activating pro-survival pathways such as the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) (Ha and Lee, 2020). DC-SIGN localizes to the cell surface of monocyte-derived immature DCs (Engering et al., 2002; Rahimi, 2021). DC-SIGN regulates transendothelial migration of DCs via ICAM-2 and the activation of resting T cells through ICAM-3 (Geijtenbeek et al., 2000). DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor nuclear factor kappa B (NF-κB) (Choi et al., 2023). Sialic acid is located in various subcellular compartments depending on the tissue type and plays critical roles in a variety of biological process (Yang et al., 2021). Sialic acid worked through mitogen-activated protein kinase (MAPK) signal (Li et al., 2023) and NF-κB pathway (Xue et al., 2018). DC-SIGN, dendritic cell-specific intercellular adhesion molecule-3 grabbing nonintegrin ICAM, intercellular adhesion molecule.
Heat shock protein 70
Heat shock protein 70 (Hsp70) exerts a critical role in several stages of ZIKV infection, including viral entry, replication, and egress. Hsp70 was also found on the cell surface, where it could interact with ZIKV during the early stages of infection, and inside the cell, where it interacts with viral RNA. Inhibiting cell surface-localized Hsp70 using anti-Hsp70 antibodies reduced ZIKV cell infection rates and generated viral particles (Fig. 2). In addition, incubating ZIKV with recombinant human Hsp70 (rhHsp70) protein reduced the amount of infectious virus in the supernatant (Pujhari et al., 2019). Confocal image analysis showed elevated Hsp70 expression in infected relative to uninfected cells and colocalization of double-stranded RNA (dsRNA) and Hsp70, suggesting that Hsp70 is a part of the virus replication complex. The reduction of virus titer in the presence of Hsp70 inhibitor (MKT077) is explained by the inhibition of viral genome transcription as a time-dependent reduction in the viral capsid mRNA was observed in virus-infected Huh7.5 cells in the presence of MKT077 (Pujhari et al., 2019). Overall, Hsp70 plays a functional role in pre- and post-ZIKV infection processes, influencing viral entry, replication, and egress (Pujhari et al., 2019).
GRP78 and ZIKV Env protein
In the co-immunoprecipitation assay, glucose-regulated proteins 78 (GRP78) was pulled down with ZIKV Env protein from human A549 cells infected with either ZIKV-T (Thai isolate) or ZIKV-U (Uganda isolate), confirming the association of ZIKV Env with GRP78 (Khongwichit et al., 2021). Antibodies against the N-terminus of GRP78 were able to impede ZIKV entry to host cells, leading to substantial decreases in ZIKV infection and viral production (Fig. 2). This phenomenon was also found after GRP78 was knocked down using short interfering RNA (siRNA), implying that GRP78 may play a role in modulating ZIKV binding, internalization, and cell replication (Khongwichit et al., 2021).
GRP78 levels were elevated during ZIKV infection and localized to locations concomitant with ZIKV Env. Also, GRP78 deletion affected ZIKV’s ability to impair host cell translation and altered the localization of viral replication factories but did not affect viral RNA synthesis. GRP78 is an important host component during ZIKV infection that may be involved in coordinating viral replication factories (Royle et al., 2020).
α2,3-Linked sialic acid
Cell surface sialic acid plays an important role in ZIKV infection. In Vero cells and hiPSC-derived NPCs, removing cell surface sialic acid with neuraminidase greatly reduced ZIKV infection (Tan et al., 2019). Moreover, knockout of the sialic acid by using the CRISPR-Cas9 gene-editing tool to delete the GNE gene showed that VeroΔGNE cells remain susceptible to ZIKV infection, but with a significant reduction of the infection efficiency of both African (Uganda-MR766) and Asian lineages (Brazil-Paraiba, French Polynesia-PF13, and Singapore-16-922) (Tan et al., 2019). Replication kinetics of ZIKV in Vero and VeroΔGNE cells imply that sialic acid is necessary for efficient ZIKV infection. ZIKV infection of VeroΔGNE cells yielded smaller plaque morphology than the Vero cells, implying that sialic acid knockout restricts ZIKV infection.
Furthermore, Huh7 cells deficient in 2,3-linked sialic acid due to ST3-galactoside-2,3-sialyltransferase 4 (huh7ΔST3GAL4) deletion had considerably lower ZIKV infection (Tan et al., 2019). The importance of sialic acid in ZIKV internalization, but not attachment, was explored using pronase. Neuraminidase-treated Vero or VeroΔGNE cells were incubated with ZIKV PF13 at 4°C or 37°C to promote virus attachment and internalization, respectively. For virus internalization, ZIKV-infected cells were subjected to pronase treatment to remove plasma membrane-bound but uninternalized viral particles after incubation at 37°C. This experiment was performed to differentiate the role of sialic acid in attachment versus internalization. ZIKV particles attached to the neuraminidase-treated and untreated cells at 4°C appeared to have no difference, confirming that sialic acid is not essential for initial attachment. However, when the cells were incubated at 37°C (to facilitate internalization) followed by pronase treatment to eliminate surface-attached but non-internalized virions, a significant reduction of the viral RNA in the neuraminidase-treated Vero cells was observed. This result indicates that ZIKV internalization is, at least in part, dependent on sialic acid (Tan et al., 2019).
Dendritic cell-specific intercellular adhesion molecule-3 grabbing nonintegrin
The DC-SIGN or CD209 is a promising receptor/attachment factor for flaviviruses. This C-type (calcium-dependent) lectin is expressed on macrophages and dendritic cells, the principal targets during dengue virus (DENV) infection in vivo (Navarro-Sanchez et al., 2003; Tassaneetrithep et al., 2003) and also targets of ZIKV infection (Hamel et al., 2015).
DC-SIGN may mediate the cell entry of ZIKV. The presence of monoclonal antibodies to various C-type lectins, competitive inhibitors rich in mannose, or antiviral gene therapy may be able to diminish ZIKV replication by interfering with the binding of viral N-glycan of E protein to DC-SIGN GAGs (Routhu et al., 2019). Cells expressing DC-SIGN, such as human dermal fibroblasts, epidermal keratinocytes, and immature dendritic cells, tolerate the most recent ZIKV isolates (Hamel et al., 2015)
Compared with parental or langerin-expressing Raji cells, DC-SIGN-expressing Raji cells readily bound ZIKV. Moreover, binding by DC-SIGN-Raji was blocked by antibodies against DC-SIGN but not by isotype antibodies. In contrast to Langerin-Raji, DC-SIGN-Raji cultured with ZIKV for 4 h successfully transferred the virus to target cells. Mannan, a carbohydrate that blocks C-type lectin receptors (CLRs) or antibodies against DC-SIGN but not the isotypes, also suppressed ZIKV transmission (Fig. 2) via DC-SIGN Raji. So, unlike langerin, DC-SIGN is implicated in ZIKV binding and transmission (Eder et al., 2023).
Clathrin-Dependent and -Independent Pathways for ZIKV Entry into the Cells
ZIKV was shown to enter human microglial (CHME3) and fibroblast (HT1080) cells via a clathrin-dependent route (Persaud et al., 2018). Chlorpromazine and pitstop2 can inhibit CME. Chlorpromazine inhibits the formation of clathrin-coated pits at the cell surface while promoting the formation of a clathrin lattice on endosomes (Wang et al., 1993). Both chlorpromazine and pitstop2 prevented ZIKV internalization into human glioblastoma cells T98G in a dose-dependent manner (Li et al., 2020). siRNA knockdown of clathrin heavy chain in T98G (Li et al., 2020) or CHME3 cells (Meertens et al., 2017) did not affect the expression of Axl but dramatically decreased the ZIKV infection. A similar phenomenon was observed in HeLa cells stably expressing Axl (HeLa-Axl), where ZIKV particles are internalized via clathrin-dependent endocytosis and then delivered to early endosomes, the mildly acidic environment, which is suitable for triggering the envelope’s conformational shift (Meertens et al., 2017).
In addition, it appears that ZIKV can also enter T98G cells through clathrin-independent pathways. The caveola-mediated route is critical for ZIKV entry into T98G cells (Li et al., 2020). It has been demonstrated that in the absence of caveolins, no caveolae are observed and that when caveolins are expressed in cells lacking caveolae, the production of caveolae is reduced (Fra et al., 1995). Both siRNA targeting caveolin-1 and the overexpression of dominant-negative caveolin-1 can inhibit caveola-mediated endocytosis, in which ZIKV entry into cells was significantly reduced (Li et al., 2020). Moreover, ZIKV colocalized with caveolin-1 in T98G cells. These findings strongly suggest that ZIKV infection of T98G cells also relies on caveola-mediated endocytosis (Li et al., 2020).
Targeting the Entry of ZIKV into the Cells as Potential Antiviral Therapeutics
Currently, there are no licensed antiviral vaccines or medicines for ZIKV infection. However, a screening assay of FDA-approved drugs in various human cell lines infected with ZIKV identified 20 compounds that significantly reduced ZIKV infection in vitro. Ivermectin, mefloquine hydrochloride, bortezomib, daptomycin, and mycophenolic acid are some of the most well-known (Barrows et al., 2016). These molecules either interfere with receptor binding or hinder internalization through endocytosis. Curcumin (Mounce et al., 2017), ZINC33683341, and ZINC49605556 (Fernando et al., 2016) inhibit viral entry by blocking receptor binding. Compounds such as chloroquine (Shiryaev et al., 2017) and suramin have also been shown to inhibit ZIKV internalization in vitro and in vivo (Tan et al., 2017).
Suramin inhibits a very early step of the replication cycle and the dispersal of infectious progeny (Albulescu et al., 2017). Binding experiments at 4°C using 35S-labeled ZIKV demonstrated that suramin interferes with virus attachment to host cells. The inhibitory effect of suramin on ZIKV attachment and virion formation, and its broad-spectrum activity, justify further evaluation of this compound as a potential therapeutic (Albulescu et al., 2017). Nanchangmycin is a polyether derived from Streptomyces nanchangensis. It inhibits the replication of ZIKV in osteosarcoma cells (U2OS) cells in vitro. Nanchangmycin targets Axl receptors and inhibiting CME (Rausch et al., 2017). Arbidol (ARB, umifenovir) was used clinically for decades as an anti-influenza virus drug (Shi et al., 2007). ARB hinders ZIKV replication in cell lines and safeguards against ZIKV-induced cytotoxic effects (Fink et al., 2018).
Since some flaviviruses that enter cells are known to be sensitive to pH changes (Oliveira et al., 2017), two drugs have been used to elevate intravascular pH to test the pH dependency of ZIKV entry (Owczarek et al., 2019). Vero cells were treated with either ammonium chloride (NH4Cl) or Bafilomycin (Baf A1) 1 hour before infection. The cells were then infected with ZIKV in the presence of NH4Cl or Baf A1 for 3 days at 37°C. Results revealed that viral particles in Baf A1-treated cells were visible in the cytoplasm, likely trapped in the endosomal hub and unable to undergo fusion. Interestingly, NH4Cl-treated cells exhibited a different distribution of the virus within cells. Only a few ZIKV virions were visible in the cytoplasm. At the same time, the majority of ZIKV particles are localized to the cell surface (Owczarek et al., 2019), demonstrating that the NH4Cl-mediated inhibition results from rewiring of the endosomal hub rather than a simple increase in endosomal pH (Owczarek et al., 2019).
Other chemical compounds have the potential to inhibit ZIKV replication by targeting viral proteins that are essential for the virus's life cycle. For example, Sofosbuvir, an FDA-approved antiviral used against the hepatitis C virus, has been shown to inhibit ZIKV RNA polymerase activity in various cell lines such as hepatoma (Huh-7), neuroblastoma (SH-SY5Y) cells, neural stem cells (NSCs), and brain organoids (Sacramento et al., 2017). (2E)-N-benzyl-3-(4-butoxyphenyl) prop-2-enamide (SBI-0090799) inhibits ZIKV infection by preventing the formation of membranous compartments in the endoplasmic reticulum (ER) and suppressing NS4A activity (Riva et al., 2021). Bafilomycin A1, an inhibitor of V-ATPase, has also shown promise as an antiviral target at the endosomal-lysosomal compartment level. It can reduce ZIKV release by interfering with viral maturation (Sabino et al., 2019). Table 1 contains more compounds tested for ZIKV infection.
Targeting the Entry of ZIKV for Antiviral Strategy
ADCC, antibody-dependent cell-mediated cytotoxicity; ZIKV, Zika virus; IMPDH, Inosine-5′-monophosphate dehydrogenase; CDKs, Cyclin-dependent kinases.
Neutralizing antibodies are critical to the protection against ZIKV infections (Robbiani et al., 2017). Neutralizing antibodies have been extensively studied against ZIKV infection (Table 1). A few specific/cross-reactive monoclonal antibodies (mAbs) were isolated from convalescent patients and immunized animals (Robbiani et al., 2017; Wang et al., 2016). Structural studies shed light on the neutralizing mechanisms of mAbs. For example, MAb C10 binds to specific residues at the intradimer interface of Env proteins. At pH6.5, C10 locks all virus surface Env proteins, and at pH5.0, it locks the Env raft structure, indicating that C10 hinders the structural rearrangement of the Env proteins during the fusion event, a crucial step in infection (Zhang et al., 2016). Similar intradimer epitopes are also observed for mAb C8 and mAb A11 (Barba-Spaeth et al., 2016).
A subset of mAbs targets the N-terminal region of ZIKV NS1. These mAbs inhibit the infection of ZIKV in different ways. MAbs 3G2 and 4B8 are more effective than mAb 4F10 in suppressing ZIKV infection in C57BL/6 neonatal mice. While mAb 4F10 primarily triggers antibody-dependent cell-mediated cytotoxicity (ADCC), mAb 3G2 and mAb 4B8 not only trigger ADCC but also inhibit ZIKV infection without Fcγ receptor-bearing effector cells, possibly at postentry stages (Yu et al., 2021). The mAbs 3G2 and 4B8 target the N-terminal region of NS1 protein, while mAb 4F10 targets the C-terminal region, implying that the effectiveness of an NS1-targeted mAb may be associated with its epitope recognition (Yu et al., 2021).
ZIKV Env resembles similarities with other known flavivirus Env structures, but it also possesses a distinct, positively charged patch next to the fusion loop region of the adjacent monomer (Dai et al., 2016). The structure of the ZIKV Env-2A10G6 complex shows that the antibody recognizes a highly conserved fusion loop and binds to ZIKV Env with high affinity and can neutralize currently circulating ZIKV strains in vitro and mice models (Dai et al., 2016). The hydrophobic residues, namely W101, L107, and F108, mediate most interactions between 2A10G6 and ZIKV Env. These residues are highly conserved among most flaviviruses, which explains the broadly neutralizing capacity 2A10G6 (Deng et al., 2011).
In another study, researchers isolated five ZIKV-neutralizing mAbs from E80-immunized mice (Qu et al., 2020). These mAbs, known as 3E8, 5F8, 5G3, 8A2, and 9C3, specifically bound to and neutralized Asian-lineage ZIKV strains. Epitope mapping revealed that all five mAbs recognized a novel linear epitope located on the glycan loop of the Env protein domain I (Qu et al., 2020). One representative of this mAb class, 5F8, was found to primarily inhibit the early stage of the postattachment viral entry process in vitro. Importantly, mAb 5F8 provided full protection in a mouse model of ZIKV lethal infection (Qu et al., 2020). These mAbs are strong candidates for developing mAb-based therapeutic drugs to treat ZIKV infection (Qu et al., 2020).
Specific human mAbs were isolated from a single patient infected with ZIKV (Wang et al., 2016). Two antibodies, namely Z23 and Z3L1, were found to possess strong ZIKV-specific neutralization abilities in vitro (Wang et al., 2016). Moreover, these antibodies were able to provide postexposure protection to mice in vivo (Wang et al., 2016). Human mAb ZIKV-117 can neutralize infection of ZIKV strains belonging to African and Asian-American lineages (Sapparapu et al., 2016). ZIKV-117 recognizes a unique quaternary epitope located at the dimer-dimer interface of the Env protein. The treatment with these mAbs significantly reduced tissue pathology, placental and fetal infection, maternal-fetal transmission, and ultimately lowering mortality rates in the mice (Sapparapu et al., 2016).
Concluding Remarks
ZIKV, an arbovirus with multiple lineages, continues to pose a global health threat, particularly in densely populated developing countries where health awareness is lacking. This virus can infect various cell types in the brain and other organs. It is crucial to sustain efforts for the prevention and clinical management of ZIKV-related diseases. However, the cell biology of ZIKV entry remains poorly understood. The virus’s ability to infect cells is linked to the exposure of negatively charged lipids (phosphatidylserine) on the viral envelope and the recognition of the viral Env protein by host receptors. Viral particles enter host cells through CME regulated by the binding of the viral protein Env. The rolling and accumulation of viral particles on the host cell surface facilitate viral entry. The expression of various binding factors on the surface of host cells determines the viral tropism. Key factors include the transmembrane receptor AXL, DC-SIGN, Tyro3, and TIM-1. These factors are vital for the viral endocytic process. Therefore, understanding ZIKV entry mechanisms aids in developing anti-ZIKV therapies targeting host cellular components. The virus enters cells through macropinocytosis, and the host ubiquitin system plays a crucial role in ZIKV tropism. These insights provide a foundation for developing targeted therapeutic interventions against ZIKV.
Footnotes
Authors’ Contributions
L.X. and N.A.H.: Conceptualization. N.A.H.: Visualization and writing. N.A.H. and L.X.: Article revision. All authors contributed to the article and approved the submitted version.
Author Disclosure Statement
The authors have declared that no competing interest exists.
Funding Information
This work was supported by the Programme of Introducing Talents of Discipline to Universities (D21004).