
Abstract
Herpesviruses are widely disseminated in the population and establish lifelong latency, which is associated with a variety of pathological consequences. A recent report showed that mice latently infected with either murine γ-herpesvirus-68 (γHV68) or murine cytomegalovirus (mCMV), mouse pathogens genetically similar to the human herpesviruses, Epstein-Barr virus, Kaposi's sarcoma–associated herpesvirus, and cytomegalovirus, had enhanced resistance to subsequent bacterial infection, suggesting protective as well as deleterious effects of latency. Here we confirm that latent γHV68 infection confers protection against subsequent infection with Listeria monocytogenes. However, the effect is transient, lasting only a few months.
Introduction
Murine gammaherpesvirus 68 (γHV68, MHV-68, or Murid herpesvirus-4) is a natural rodent pathogen that is structurally, biologically, and genetically related to the human γ-herpesviruses (4,15,25). Intranasal infection of mice with γHV68 causes an acute respiratory infection that is rapidly resolved, followed by the establishment of a lifelong latent infection. Because of its close homology to human γ-herpesviruses, γHV68 provides a valuable in vivo experimental model to study γ-herpesvirus pathology and host immunity.
A recent report has described symbiotic protection against subsequent bacterial challenge in mice harboring latent β-and γ-herpesvirus infections (2). Specifically, it was shown that mice latently infected with either the β-herpesvirus, murine cytomegalovirus (mCMV), or the γ-herpesvirus γHV68, were protected against lethal challenges by Listeria monocytogenes or Yersinia pestis. Protection was also manifest by a reduction in the numbers of bacteria recovered from the spleens and livers of latently infected mice following challenge. These protective effects were restricted to the latent phase of infection and induction of protection was virus-specific, as mice previously infected with the α-herpesvirus HSV-1 or Sindbis virus failed to exhibit increased resistance against similar bacterial challenges. Host protection was observed in latently infected mice as far out as 3 mo post-infection, and was shown to be associated with elevated serum levels of inflammatory cytokines including IFN-γ and TNF-α, which are proposed to result in the chronic activation of bactericidal macrophages. While investigating the impact of γHV68 infections on immunity in aged mice, we discovered that the symbiotic protective effect failed to persist with lifelong latency. This observation prompted us to determine how long after the establishment of γHV68 latency this symbiotic protection persisted.
Materials and Methods
Mice
Female C57BL/6 mice were obtained from the Trudeau Institute animal breeding facility. The mice were housed under specific pathogen-free conditions before infection, and in ABSL3 containment after viral infection. The Institutional Animal Care and Use Committee at the Trudeau Institute approved all animal procedures.
Viral infection
γHV68, clone WUMS (Washington University School of Medicine), was propagated and titered on NIH-3T3 fibro-blasts (ATCC CRL1568) (25). Mice between 6 and 8 wk of age were anesthetized with 2,2,2-tribromoethanol and intranasally infected with 400 PFU of γHV68. Mice were considered latently infected at 30 d after the initial γHV68 infection. Viral latency was confirmed with quantitative PCR using primers specific for the γHV68 ORF50 gene on DNA isolated from the spleens of infected mice (24).
Bacterial infection and measurement of bacterial loads
Stocks of L. monocytogenes (strain EGD, supplied by Robert North, Trudeau Institute) were prepared after passage through C57BL/6 mice (12). The mice were challenged with 2.5 × 106 cfu of bacteria via intraperitoneal injection, and spleens and livers were removed from infected mice 3 d later. The numbers of viable bacteria in each tissue type was determined by plating tissue homogenates on BHI-agar plates.
Measurement of cytokine levels
Cytokines were detected in whole sera taken from γHV68-infected mice at the indicated times post-infection using the mouse inflammation Cytometric Bead Array (BD Biosciences, San Jose, CA) according to the manufacturer's instructions.
Data analysis
Statistical significance was calculated using the nonparametric Mann-Whitney rank test using Prism 4 software (GraphPad Software, La Jolla, CA). p Values <0.05 were considered significant.
Results
γ-Herpesvirus latency-mediated protection against bacterial infection is short-lived
Previous studies have described a symbiotic protection from bacterial infection in mice that harbor a latent β- or γ-herpesvirus infection that persisted for up to 3 mo post-infection (2). To determine how long after infection this protection was maintained, we challenged mice latently infected with γHV68 with L. monocytogenes at periods between 2 and 19 mo after the initial viral infection. Consistent with the previous findings we observed that during the first 5 mo following γHV68 infection, latently-infected mice exhibited greatly reduced viable bacteria counts in spleen and liver tissue as compared to age-matched uninfected control mice (Fig. 1A and B). However, this protective effect was completely lost in mice latently infected with γHV68 after more than 5 mo, in that as early as 6.5 mo after infection mice were found to have viable bacteria counts similar to age-matched uninfected controls, indicating that latency-induced host protection waned with time. Importantly, the viral load assessed in terms of genome copy was constant between the 5- and 6.5-mo time points (Fig. 1C). These results indicate that the enhanced resistance against bacterial infection conferred to the host by γ-herpesvirus latency is finite and not maintained throughout lifelong viral latency.
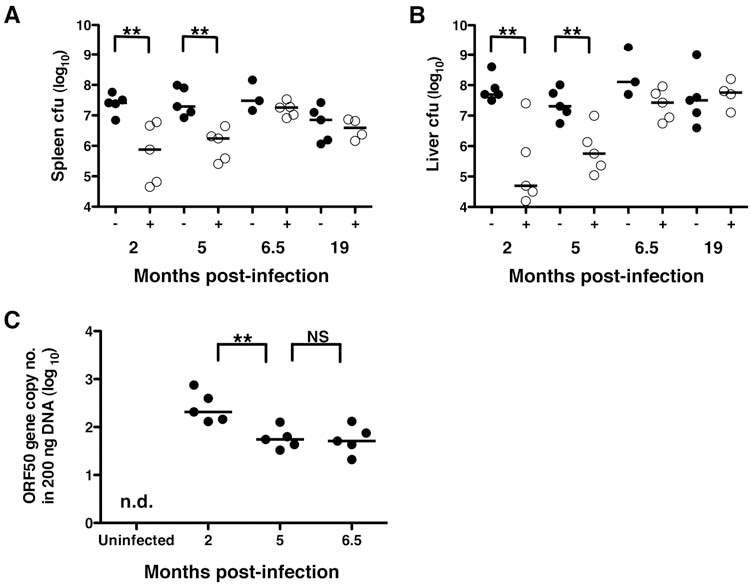
Herpesvirus latency-mediated protection against subsequent bacterial infection is finite. Mice latently infected with γHV68 for the indicated lengths of time (indicated by “+” and open circles) and uninfected age-matched control mice (indicated by “−” and closed circles) were challenged with L. monocytogenes. The number of viable bacteria detected in the spleens (
Transient elevation in the levels of inflammatory cytokines following the establishment of γ-herpesvirus latency
It was proposed that the enhanced resistance to bacterial pathogens observed in latently infected mice was due to a systemic activation of macrophages and heightened innate immunity, likely due to a prolonged and elevated production of inflammatory cytokines (2). Consistent with this notion, the authors found that levels of TNF-α and IFN-γ remained elevated in the serum of latently infected mice for up to 41 d following γHV68 infection. To determine how long IFN-γ and TNF-α cytokine levels remained elevated in the serum of latently infected mice, we measured the cytokine levels in sera of γHV68-infected mice at extended times following the initial infection. Consistent with the original report (2), elevated levels of IFN-γ (Fig. 2A) and TNF-α (Fig. 2B) were detected in the sera of mice 30 d following γHV68 infection. However, the levels of these two cytokines returned to baseline in latently infected mice at 2 mo post-infection, where they remained for as long as 8 mo post-infection. These results indicate that γHV68 infection increases host production of cytokines known to influence macrophage activation and pathogen resistance; however, this effect is apparently lost shortly after the establishment of latency.
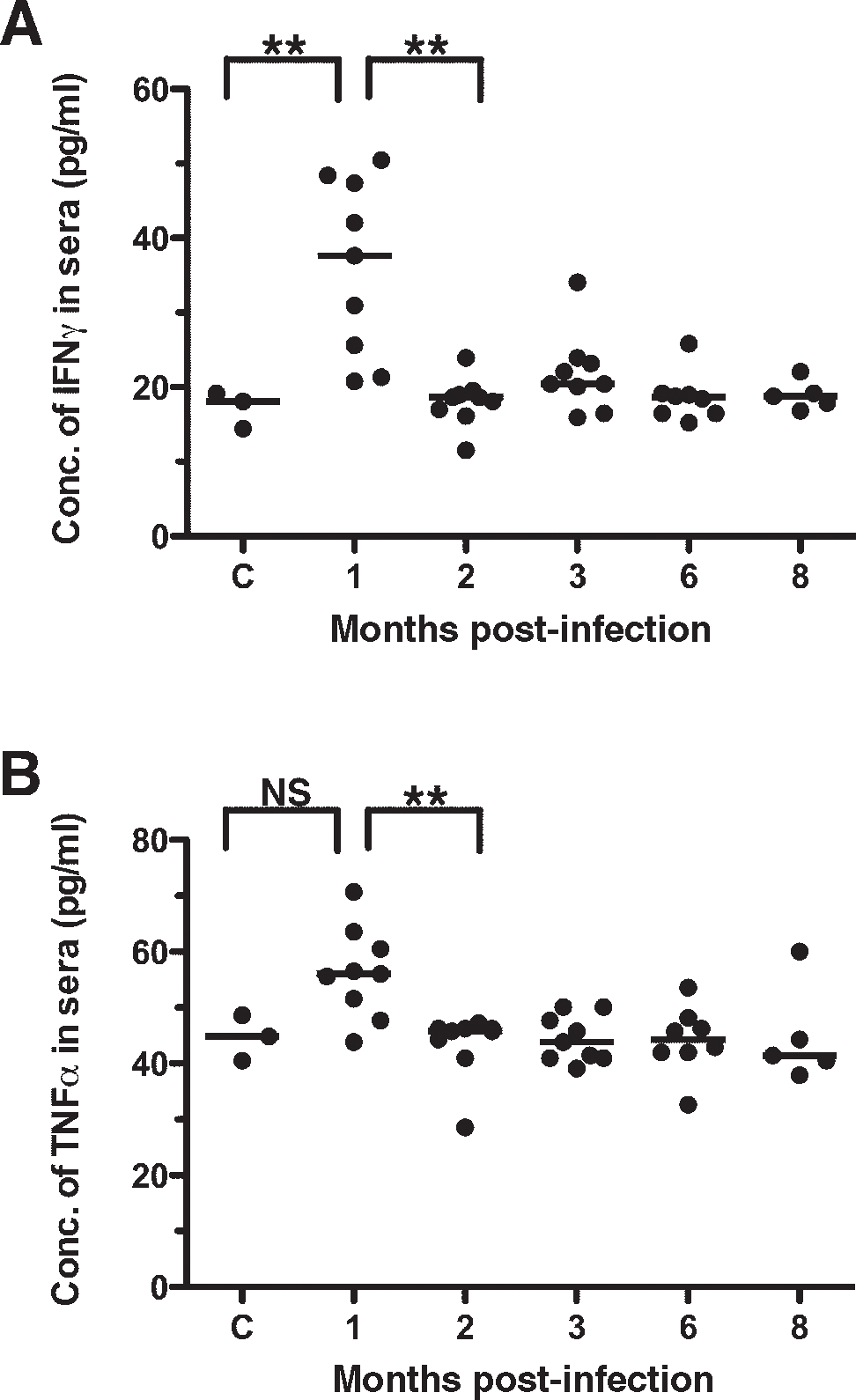
Temporary elevation in the levels of inflammatory cytokines following the establishment of γ-herpesvirus latency. The levels of IFN-γ (
Discussion
Our results confirm the findings of a previous study that demonstrated that herpesvirus latency conferred host protection against subsequent bacterial infection (2). The authors speculated that latent herpesvirus infection represented a symbiotic relationship in which heightened activation of the innate immune system protects the host from ensuing infections. These data raised the possibility that latent herpesvirus infection may in fact be beneficial to the host, and suggested that vaccines to prevent or control latency may have unintended negative consequences for host immunity. Here we confirm that beneficial effects do exist, but this protection is transient, lasting only as long as 5 mo following acute γHV68 infection, despite a stable viral load. Furthermore, elevated serum levels of IFN-γ and TNF-α, which were associated with the protective effect, declined even more quickly and could no longer be detected at 2 mo after the initial infection. Thus, rather than representing symbiosis, latency-mediated protection is likely a consequence of transient changes in the activation status and host immunity responsible for the initial reduction of the number of virus-infected cells and establishment of latency.
IFN-γ was shown to be critical for the symbiotic protection against bacterial infection, with elevated levels in the serum for as long as 41 d post-infection, and latently-infected IFN-γ-deficient mice were not protected from bacterial infection (2). IFN-γ has been shown to block latent γHV68 reactivation in a cell type–specific manner via regulation of viral latent gene expression (21,22). Both CD4 and CD8 T cells produce IFN-γ during γHV68 latency (3). In addition, it was recently shown that the γHV68 M1 gene product induces persistent activation of CD8 T cells bearing a Vβ4 T-cell receptor, and that these T cells appear capable of suppressing virus reactivation via long-term IFN-γ production (6). Although IFN-γ is produced at high levels following the initiation of the host antiviral immune response, it is not critical for control of the initial γHV68 infection (19). However, IFN-γ appears to be critical for long-term control of γHV68 infection and its symptoms, since infected IFN-γ-receptor-deficient mice show multiple organ fibrosis and enhanced virus reactivation (5,7,14,23). Why then are the elevated levels of IFN-γ seen early after γHV68 infection not maintained during latency? We speculate that as the level of latent virus equilibrates, the amount of viral antigen is reduced, and as a consequence fewer virus-specific T cells are induced to produce IFN-γ and other cytokines. Thus, sufficient levels of IFN-γ are still being produced to control virus reactivation, but not enough to maintain protection against ensuing bacterial challenges.
Although γ-herpesvirus infections may provide the human host a temporary benefit of protection against subsequent infections as observed in the mouse, the transient protection afforded by herpesvirus latency is unlikely to outweigh the deleterious consequences of infection. Although the pathogens are widely distributed in the human population, generally there is low incidence of disease. Nevertheless, there are specific susceptibility groups with high risk for pathological consequences. For example, there is a high incidence of endemic Burkitt's lymphoma in equatorial Africa (9,13), and there are regions of Southeast Asia with a 20-fold higher than normal incidence of EBV-associated nasopharyngeal carcinoma (11). Immunosuppression as a consequence of disease (AIDS) or post-transplant therapy also can lead to increases in EBV-associated lymphoproliferative syndromes and lymphomas, as well as KSHV-associated Kaposi's sarcoma (16,20). Finally, there is an elevated incidence of infectious mononucleosis in developed countries, which has been associated with the development of Hodgkin's lymphoma and multiple sclerosis (8,10). Thus the development of prophylactic and therapeutic vaccination strategies against human γ-herpesviruses remains an important goal.
Conclusion
These data confirm the previous report of Barton et al. (2) that early γHV68 latency confers protection against bacterial infection, but show that the effect is transient. Therefore, there is no evidence that γ-herpesvirus latency provides a lifelong benefit to the host.
Footnotes
Disclosure Statement
No conflicting financial interests exist.
Acknowledgements
We would like to thank Drs. David Woodland and Mike Freeman for helpful discussions and comments on the manuscript. This work was supported by National Institutes of Health grants AG21600, AI51602, and AI42927 to M.A.B., and AI61577 to S.T.S. and the Trudeau Institute.