
Abstract
SHP-1 is a protein tyrosine phosphatase that negatively regulates cytokine signaling and inflammatory gene expression. Mice genetically lacking SHP-1 (me/me) display severe inflammatory demyelinating disease following intracranial inoculation with the BeAn strain of Theiler's murine encephalomyelitis virus (TMEV) compared to infected wild-type mice. Furthermore, SHP-1-deficient mice show a profound and predominant infiltration of blood-derived macrophages into the CNS following intracerebral injection of TMEV, and these macrophages are concentrated in areas of demyelination in brain and spinal cord. In the present study we investigated the role of SHP-1 in controlling CNS inflammatory demyelination following a peripheral instead of an intracerebral inoculation of TMEV. Surprisingly, we found that while wild-type mice were entirely refractory to intraperitoneal (IP) infection by TMEV, in agreement with previous studies, all SHP-1-deficient mice displayed profound macrophage neuroinvasion and macrophage-mediated inflammatory demyelination. Moreover, SHP-1 deficiency led to increased expression of inflammatory molecules in macrophages, serum, and CNS following IP infection with TMEV. Importantly, pharmacological depletion of peripheral macrophages significantly decreased both paralysis and CNS viral loads in SHP-1-deficient mice. In addition, peripheral MCP-1 neutralization attenuated disease severity, decreased macrophage infiltration into the CNS, and decreased monocyte numbers in the blood of SHP-1-deficient mice, implicating MCP-1 as an important mediator of monocyte migration between multiple tissues. These results demonstrate that peripheral TMEV infection results in a unique evolution of macrophage-mediated demyelination in SHP-1-deficient mice, implicating SHP-1 in the control of neuroinvasion of inflammatory macrophages and neurotropic viruses into the CNS.
Introduction
In support of the role for macrophages in MS, infiltrating macrophages have been demonstrated to be essential effectors of demyelination in both Theiler's murine encephalomyelitis virus (TMEV) (6,23,75,77) and experimental autoimmune encephalomyelitis (EAE) (25,40,42,87) animal models of MS. These findings have stimulated intense interest in the function of macrophages in lesion formation, including signaling events that draw these cells into the CNS white matter and trigger the effector mechanisms by which these cells damage myelin. For instance, macrophages have been identified as the major responders to CNS chemokines and producers of a number of pro-inflammatory cytokines, chemokines, and toxic molecules known to promote demyelination (29,37,57,74,78,79,90).
Several studies demonstrated that either peripheral or CNS-infiltrating macrophages from MS patients display enhanced activation profiles that could be important in their demyelinating activity (12,20,34). Induction of an increased inflammatory profile of macrophages has generally been ascribed to signals originating from autoreactive lymphocytes in MS. However, intrinsic defects in innate macrophage-mediated effector functions may also be critical to achieve substantial inflammatory potential to cause demyelinating activity in the CNS. Interestingly, we have recently demonstrated that monocyte-derived macrophages of MS patients have deficient levels of the phosphatase SHP-1 relative to those of normal control subjects (20). SHP-1 is a protein tyrosine phosphatase with two SH2 domains which acts as a negative regulator of both innate and acquired immune cytokine signaling via inflammatory transcription factors NF-κB, STAT1, and STAT6 (11,28,36,45,61,62). Accordingly, we showed that deficient levels of SHP-1 in MS macrophages were directly responsible for the increased activation of these inflammatory transcription factors compared to those of normal subjects (20).
To further elucidate the function of SHP-1 in inflammatory demyelinating disease, we have been utilizing the TMEV-induced demyelinating disease model of MS (13,17,26,30,49). When susceptible strains of mice are inoculated intracerebrally (IC) with TMEV the mice develop immune-mediated demyelinating disease resembling human MS. TMEV infection leads to biphasic disease with an early gray matter infection followed with later progressive white matter disease involving a complex array of leukocytes and pro-inflammatory molecules that eventually cause demyelination. Chronic disease is associated with limb paralysis, demyelinating spinal cord lesions, and extensive white matter mononuclear cell infiltrates, consisting primarily of lymphocytes and macrophages (50,68). It is now well accepted that while lymphocytes may act as initiators/perpetuators of demyelination, macrophages are the major effectors of demyelination that may act in large measure autonomously of lymphocytic influences (7,22,50,76,77,80).
SHP-1-deficient mice do not display the typical biphasic TMEV-induced disease seen in wild-type mice following IC inoculation, but in contrast exhibit an acute white matter disease in which inflammatory demyelination occurs very early (within the first week of infection). Moreover, in distinction to TMEV-induced demyelinating disease in wild-type mice, in which both acquired and innate immunity play an important role, TMEV-induced demyelination in SHP-1-deficient mice is mediated by blood-borne macrophages that invade the CNS (18).
Because SHP-1-deficient mice are highly susceptible to macrophage-mediated demyelination following intracranial TMEV infection(18), we asked whether these mice were similarly more susceptible than wild-type mice to peripheral TMEV inoculation with respect to both CNS infection and macrophage-mediated inflammatory demyelination. We demonstrate that following intraperitoneal (IP) infection with TMEV (BeAn strain), a high percentage of me/me mice show paralysis within the first week of infection, in sharp contrast to wild-type mice, which are completely resistant to disease. Furthermore, the spinal cords of me/me mice following IP TMEV inoculation show extensive inflammatory demyelinating lesions associated with abundant and exclusive infiltration of macrophages. Importantly, depletion of peripheral macrophages delayed the onset of TMEV-induced paralysis, decreased CNS macrophage infiltration and demyelination, and decreased CNS viral loads. Invading macrophages contained abundant viral antigens, suggesting that SHP-1 controlled both migration of monocytes from the periphery into the CNS, and macrophage infection within the CNS. Consistent with this hypothesis, we have identified the chemokine MCP-1 as an important stimulus of macrophage migration and virus infection in the CNS. These results indicate that a deficiency in SHP-1 establishes a permissive state in which peripheral TMEV infection results in enhanced macrophage infiltration into the CNS, virus replication, and inflammatory demyelination. Thus SHP-1 appears to alter multiple aspects of macrophage biology that control virus replication, chemokine responsiveness, and inflammatory activity, that directly relate to virus-induced demyelinating disease.
Materials and Methods
Animals
SHP-1-deficient motheaten (me/me) mice (C3HeB/FeJLe-a/a background) and their phenotypically normal wild-type littermates were produced from heterozygous breeding pairs obtained from Jackson Laboratories (Bar Harbor, Maine). Strain designations for heterozygous breeders are C3FeLe.B6-a/a Hcphme /+ (stock no. 000225) for motheaten mice.
Paralysis scores
Mice were scored daily for signs of paralysis. They were scored on a five-point scale: 1 = incomplete hindlimb paralysis (dragging hindlimb but able to move it); 2 = complete paralysis of one hindlimb; 3 = complete paralysis of one hindlimb and one forelimb or paralysis of both hindlimbs; 4 = quadriplegia; and 5 = death following paralysis.
Virus inoculation
The attenuated strain of TMEV, BeAn 8386, (ATCC, Manassas, VA) was propagated and titrated by plaque assay in BHK-21 cells. Cultured mouse macrophage cultures were inoculated with 1 × 106 PFU/mL at a multiplicity of infection (MOI) of 1.0. Twelve-day-old mice received a single IP injection of 1 × 106 PFU of BeAn in a volume of 0.1 mL unless otherwise specified. Mice were observed daily for paralysis. At 6 or 24 d post-infection (dpi) mice were anesthetized, blood was drawn, and the mice were perfused. Cerebral hemispheres and spinal cords were suspended in RNA STAT-60 (Tel-Test, Friendswood, TX) for RNA analysis. Also, brain and spinal cords were used to prepare single-cell suspensions for flow cytometry analysis.
In-vivo depletion of macrophages
To deplete macrophages in wild-type and me/me mice, liposome-encapsulated clodronate (clodronate liposomes) were used. Clodronate was purchased from Roche Pharmaceuticals (Germany) and was encapsulated into liposomes by Encapsula Nanosciences (Nashville, TN). The final solution contained 5 mg/mL of encapsulated clodronate. Control liposomes contained phosphatidylcholine and cholesterol without clodronate. Mice were injected IP with 0.2 mg clodronate at 2 d before infection with the TMEV virus. This injection route and the method was previously shown to specifically deplete CD11b+Ly6Chi monocytes/macrophages in multiple tissues and blood (8,71,94). As controls, mice were injected IP with the same volume of control liposomes 2 d before TMEV infection. Mice further received either control liposomes or 0.1 mg clodronate liposomes at days 2 and 6 post-infection. Macrophage depletion in clodronate-liposome-treated mice was verified by staining for CD45hiCD11b+, CD11b+Ly-6Chi, and F4/80+ cells in the spleens and CNS of mice 3 or 9 d after the first clodronate injection and was compared to mice that received the control liposome injections.
In-vivo MCP-1 neutralization
Mice received 60 μg of either control IgG or anti-MCP-1 antibodies (R&D Systems, Minneapolis, MN) on the day of IP infection with 106 pfu BeAn/mouse. Thereafter, the mice received 30 μg of control IgG or anti-MCP-1 antibodies every 2 d for 10 d. The antibody was verified that neutralized the chemoattractive function of MCP-1 in vitro (18).
Real-time RT-PCR
Total RNA was isolated using RNA STAT-60. RNA was quantified spectrophotometrically, and 0.5 μg of total RNA was converted into cDNA. Briefly, total RNA and random primers (Invitrogen, Carlsbad, CA) were incubated at 72° for 10 min. Reverse transcription was performed using the Superscript II RT enzyme (Invitrogen), according to manufacturer instructions. cDNA was diluted to 200 μL with water, and 4 μL was used for quantitative real-time RT-PCR using the SYBR Green kit (Abgene, Epsom, U.K.). The PCR parameters were 15 min at 95° and 35 cycles at 95° for 15 sec and 60° for 1 min in the ABI Prism 700 (Applied Biosystems, Foster City, CA). The primers were used at 10 nM. Serial dilutions of cDNA containing a known copy number of each gene were used in each quantitative PCR run to generate a standard curve relating copy number with threshold amplification cycle (19). Gene expression levels were calculated during the logarithmic amplification phase by determining the initial mRNA copy number using the standard curve. Amplification of each gene-specific fragment was confirmed both by examination of melting peaks and by agarose gel electrophoresis. The primer pairs used in this study were: TMEV, forward: TGGTCGACTCTGTGGTTACG, reverse: GCCGGTCTTGCAAAGATAGT; MCP-1, forward: GTATGTCTGGACCCATTCCTTC, reverse: GCTGTAGTTTTTGTCACCAAGC; TNF-α, forward: TGAACTTCGGGGTGATCGGTC, reverse: AGCCTTGTCCCTTGAAGAGAAC; IL-6, forward: CAGAGGATACCACTACCAACAG, reverse: TCTCATTTCCACCACGATTTCCC; VCAM-1, forward: CCCCAAGGATCCAGAGATTCA; reverse: ACTTGACCGTGACCGGCTT; and GAPDH, forward: ACCACCATGGAGAAGGC, reverse: GGCATGGACTGTGGTCATGA.
Flow cytometry
Characterization and quantification of immune cell infiltration into the CNS
The brain and the spinal cord were removed from mice and placed in 2 mL of cold Hank's balanced salt solution (HBSS). The tissue was homogenized with a fine-tip glass pipette and then filtered through a 40-μm mesh cell strainer cap as previously described (20,51) but without collagenase digestion. The single-cell suspensions were washed twice with cold HBSS with 5% FBS. Aliquots of 1 × 106 cells were resuspended in a 100 μL of HBSS and incubated with 10 μL of CD45-FITC, CD3-APC, CD11b-PE, CD19-FITC, CD49b-PE, and Ly-6C-FITC (Becton Dickinson, Mountain View, CA).
Intracellular staining
Single-cell suspensions in 1.0 mL PBS received 100 μL of 16% stock paraformaldehyde for fixation (1.5% paraformaldehyde) for 5 min and then incubated in 90% methanol at 4°C for 30 min to permeabilize cells for intracellular staining. The cells were washed twice with the staining media containing 0.5% BSA and 0.02% sodium azide in PBS. The levels of several intracellular antigens were concurrently analyzed with CD45-FITC. Fixed and permeabilized cells were incubated overnight at 4°C with 1 μg of goat anti-ADAM8 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) or goat polyclonal IgGs for isotype control. Then the cells were incubated for 3 h in 1 μg of swine anti-goat secondary antibody conjugated to PE (Invitrogen). Similarly, cells were single stained with rat anti-mouse LT-α (R&D Systems, Minneapolis, MN), rat anti-mouse F4/80 (Serotec, Raleigh, NC), and mouse anti-arginase-I (BD Biosciences, San Jose, CA) overnight, and then stained with swine anti-rat or goat anti-mouse secondary antibodies conjugated to PE (Invitrogen). Single-cell suspensions were also double stained with CD45 and rabbit anti-TMEV Ab (a gift from Howard L. Lipton, Northwestern University), followed by goat anti-rabbit secondary antibody conjugated to PE (Invitrogen). Cells were analyzed on an LSRII analyzer (Becton Dickinson) and the percentage of positively stained cells and the mean fluorescence intensity (MFI) was recorded. Data were analyzed with FlowJo software (Tree Star Inc., Ashland, OR).
Immunohistochemical analysis
Spinal cords from wild-type and me/me mice before and 6 days after infection with TMEV were used for immunohistochemical analysis. Spinal cords of mice that were pretreated with control liposomes or clodronate liposomes and then infected with TMEV were also analyzed. Mice were anesthetized and intracardially perfused via the left ventricle, first with 10 mL PBS, followed by 20 mL of 4% paraformaldehyde in PBS. Spinal cords were further incubated in 4% paraformaldehyde in PBS for 1 h and then dissected out and placed in cold 30% sucrose in PBS overnight. Spinal cords were embedded in OCT compound, frozen on dry ice, and sectioned with a cryostat at 8-μm thickness at −16°C. To double stain with MBP and CD11b, sections were first stained for mouse CD11b mAb directly conjugated to biotin (R&D Systems), followed by incubation in a streptavidin-alkaline phosphatase conjugate. A blue alkaline phosphatase reaction product was produced using a BCIP/NBT substrate kit (Zymed/Invitrogen). The same sections were then stained with a goat anti-MBP Ab (sc-13914; Santa Cruz Biotechnology), followed by anti-goat immunoglobulin-HRP (Dako, Carpinteria, CA), and red color was developed using the HRP substrate AEC chromogen (Zymed/Invitrogen). Spinal cords also were similarly double stained for MBP as above, and with rabbit anti-TMEV Ab (gift from Howard L. Lipton, Northwestern University), followed by HRP-swine anti-rabbit secondary antibody. (Invitrogen). Also, the spinal cords were stained with rat biotinylated anti-CD4 and anti-CD8 monoclonal antibodies (Biosource, Carlsbad, CA).
Macrophage culture
For the macrophage cultures, spleens were removed from mice and ground between the frosted surfaces of two glass slides in HBSS. Freed cells were left for 2 min on ice, pelleted by centrifugation, and the supernatant discarded. The pellet was resuspended in 4 mL/spleen of red blood cell lysis buffer (155 mM NH3Cl, 0.1 mM EDTA, and 12 mM NaHCO2) and incubated on ice for 1 min. The cells were washed twice with HBSS and were resuspended in RPMI medium with 15% FBS and supplemented with 10% (v/v) L929 cell (ATCC) culture supernatant (sL929-medium) as a source for colony-stimulating factor (CSF-1) (10). The cells were plated at a density of 5 × 105 cells/mL and were fed every 3 d. On day 9, viability was evaluated with trypan blue and the adherent cells were infected with the TMEV virus for 48 h. Following culture, more than 97% of the cells were CD11b+ Ly6C+ as assessed by flow cytometry. In addition, bone marrow–derived macrophages were derived from single-cell suspensions of femoral bone marrow cultured in 15% FBS and supplemented with 10% (v/v) L929 cell culture supernatant for 1 wk.
Chemokine/cyokine ELISA
The levels of the cytokines TNF-α, IL-1β, and IL-6, and the chemokine MCP-1 were quantified in serum and brain homogenates. Brains were homogenized in RIPA buffer, the amount of total protein was quantified, and the amount of TNF-α and IL-6 was quantified using DuoSet ELISA kit (R&D Systems) following the manufacturer's protocol.
Statistical analysis
Histograms or tables represent the mean values with standard errors. The number of samples per individual mouse used in each assay is indicated in the figure legend. The p values were generated using the unpaired Student's t-test and a p value <0.05 was used for statistical significance.
Results
Peripheral TMEV inoculation causes paralysis in SHP-1-deficient mice
Previously we have shown that intracranial inoculation of the attenuated BeAn strain of TMEV resulted in a rapid and severe paralytic demyelinating disease in SHP-1-deficient mice (60). Furthermore, in SHP-1-deficient mice the severity of paralysis, CNS viral loads, and demyelination were all dependent on the infiltration of peripheral macrophages into the CNS (18). Because of the profound susceptibility of me/me mice to TMEV and the inherently unnatural route of infection used in these prior studies, it was of interest to determine whether me/me mice may also be susceptible to CNS disease following peripheral inoculation of TMEV. Therefore, wild-type and SHP-1-deficient mice (me/me) mice received IP inoculation with different doses of BeAn TMEV (Fig. 1). The mice were further monitored for signs of paralysis for a period of 3 mo and the numbers of mice that showed signs of paralysis were determined (Fig. 1B). SHP-1-deficient mice that received either 108 or 106 pfu of TMEV showed initial signs of paralysis at approximately 3 dpi, and by 6 dpi almost all of the mice were at 5 on our severity scale (Fig. 1A). Furthermore, the amounts of virus injected correlated with disease severity in me/me mice. In sharp contrast wild-type mice did not show any evidence of paralysis, even when infected with 108 pfu and observed for 3 mo. We concluded that a genetic deficiency in SHP-1 in mice allowed rapid progression of peripheral TMEV infection to clinical signs of paralysis.
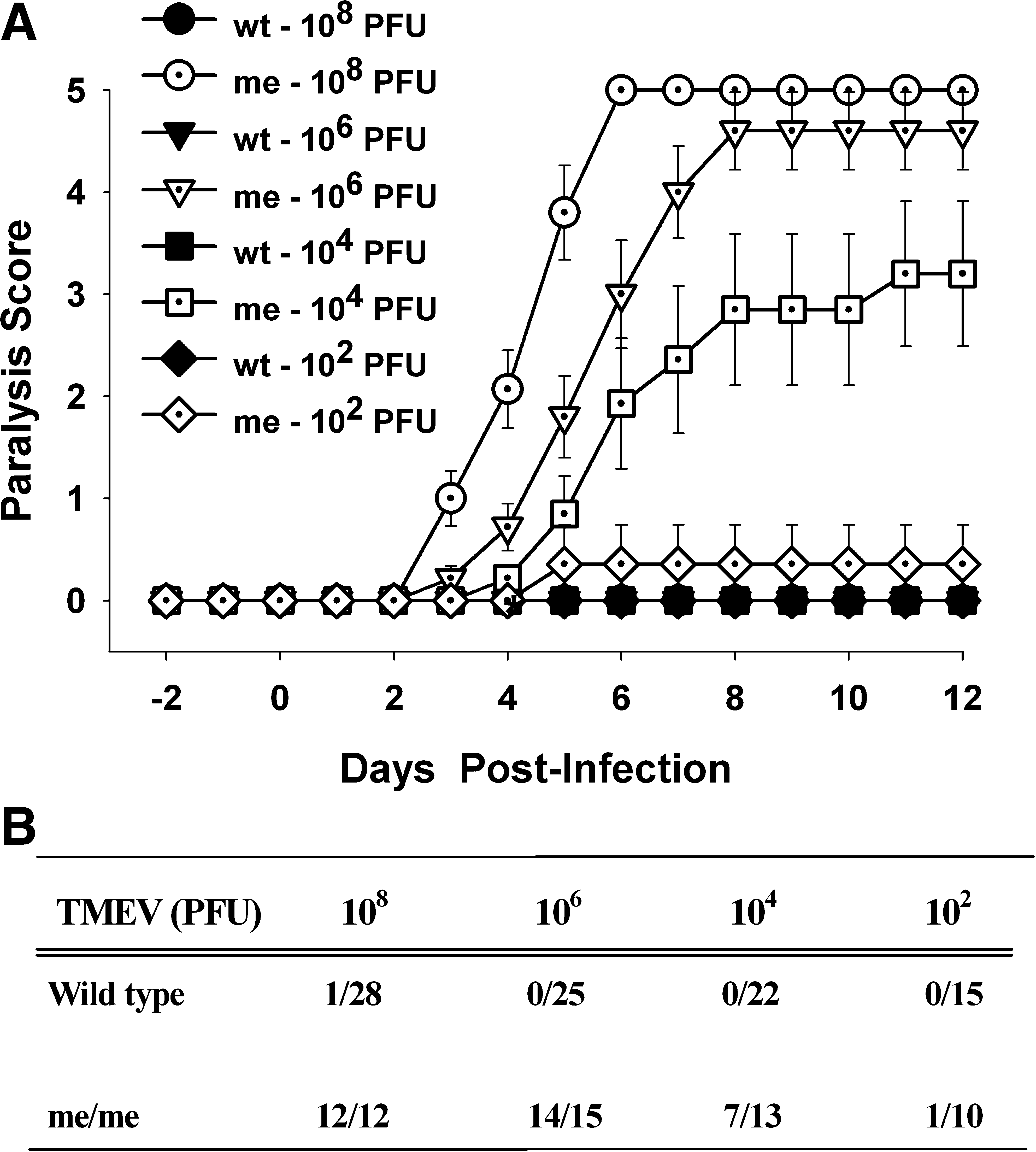
Paralysis scores of wild-type (wt) and SHP-1-deficient (me) mice following IP injection with different doses of BeAn TMEV. Mice were infected with 108, 106, 104, and 102 pfu/mouse and were observed daily for paralysis and scored on a five-point scale as described in the text. (
Peripherally-inoculated SHP-1-deficient mice display massive macrophage infiltration at sites of inflammatory spinal cord demyelination
Next it was important to determine the likely inflammatory correlates of CNS clinical signs in infected SHP-1-deficient mice (Fig. 2). Spinal cords of wild-type and me/me mice were homogenized into single-cell suspensions before and at 1 and 6 dpi with 106 pfu of TMEV, and the presence of blood-derived leukocytes was quantified using flow cytometry as previously described (18,51). First, the cells were double stained with CD11b and Ly6C, recently established as a reliable marker of blood-derived macrophages (1,83,84) (Fig. 2A and B). At 24 h dpi there was a small, insignificant increase in CD11b+Ly6Chi inflammatory macrophages in the spinal cords of me/me mice. At 6 dpi there was a significantly higher number of CD11b+Ly6Chi macrophages in the spinal cords of me/me mice, in sharp contrast to wild-type mice (Fig. 2A and B). Furthermore, the infiltrating macrophages in infected me/me mice stained positive for CD45hiCD11b+ and F4/80+ (data not shown), as previously described (18). To directly address whether other leukocyte subsets invaded the CNS following infection, we stained tissue homogenates with antibodies to CD3, CD49b, and CD19, and determined that the levels of T, NK, and B cells, respectively, did not significantly increase in the CNS following TMEV infection in either wild-type or me/me mice (data not shown).
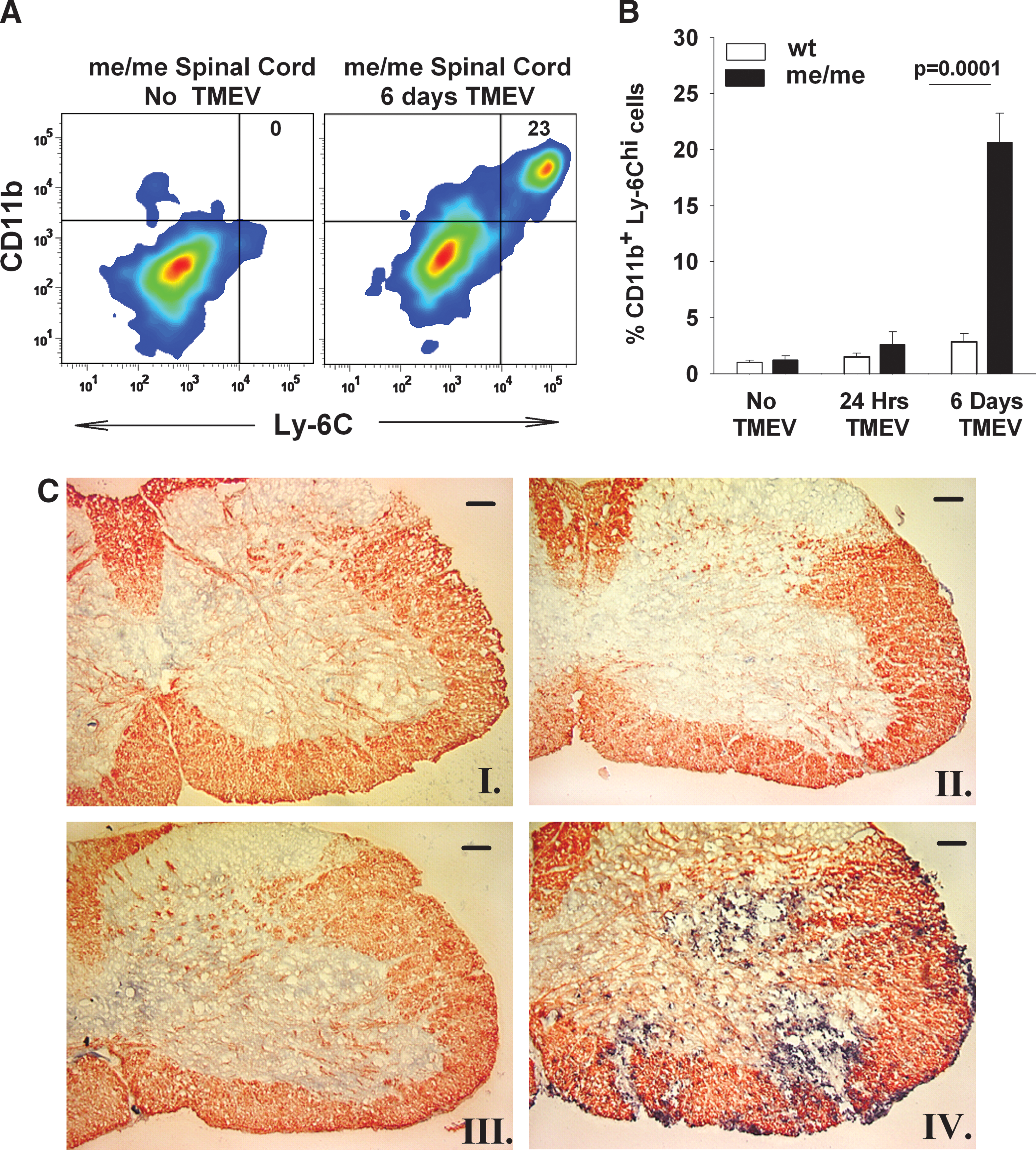
Immune cell infiltration into the spinal cords of wild-type (wt) and SHP-1-deficient mice (me) mice following IP injection of 106 pfu BeAn/mouse. Single-cell suspensions of spinal cords of wt and me/me mice were stained with CD11b and Ly-6C at 6 d post-TMEV IP injection. Stained whole-cell suspensions were assayed with flow cytometry and the percentage of CD11b+Ly-6Chi cells was recorded. (
In order to further document that macrophages are the main inflammatory cellular infiltrate seen in the CNS of paralyzed me/me mice infected IP with TMEV, we examined the localization of these cells in the spinal cord sections by microscopy. Spinal cords from wild-type and me/me mice were fixed, frozen, and sectioned, and stained by double-color immunohistochemistry of CD11b and myelin basic protein (MBP) before and at 6 d post-TMEV infection. The spinal cords of each group of animals (at least four animals per group) were surveyed in several sections at different levels of the spinal cord and representative sections are shown (Fig. 2C). Such staining allowed an examination of the spatial distribution between macrophage infiltration (CD11b accumulation) and demyelination (MBP loss from specific white matter tracts). TMEV-infected wild-type mice did not show focal demyelination or presence of CD11b+ cells (Fig. 2C, panel II). In contrast, the spinal cords of infected me/me mice displayed extensive areas of demyelination, primarily in ventrolateral tracts that contained large numbers of CD11b+ cells concentrated within the lesions (Fig. 2C, panel IV). Focal demyelination associated with macrophage infiltration was seen throughout the white matter of spinal cords of infected me/me mice, with more severe lesions observed in the lumbar section. Higher magnification revealed that the CD11b+ cells displayed a simple rounded morphology consistent with blood monocyte origin. In contrast, spinal cords of infected wild-type and me/me mice stained with anti-CD4 and anti-CD8 antibodies showed no immunoreactivity (data not shown).
Increased viral loads and inflammation in the CNS of peripherally-inoculated SHP-1-deficient mice
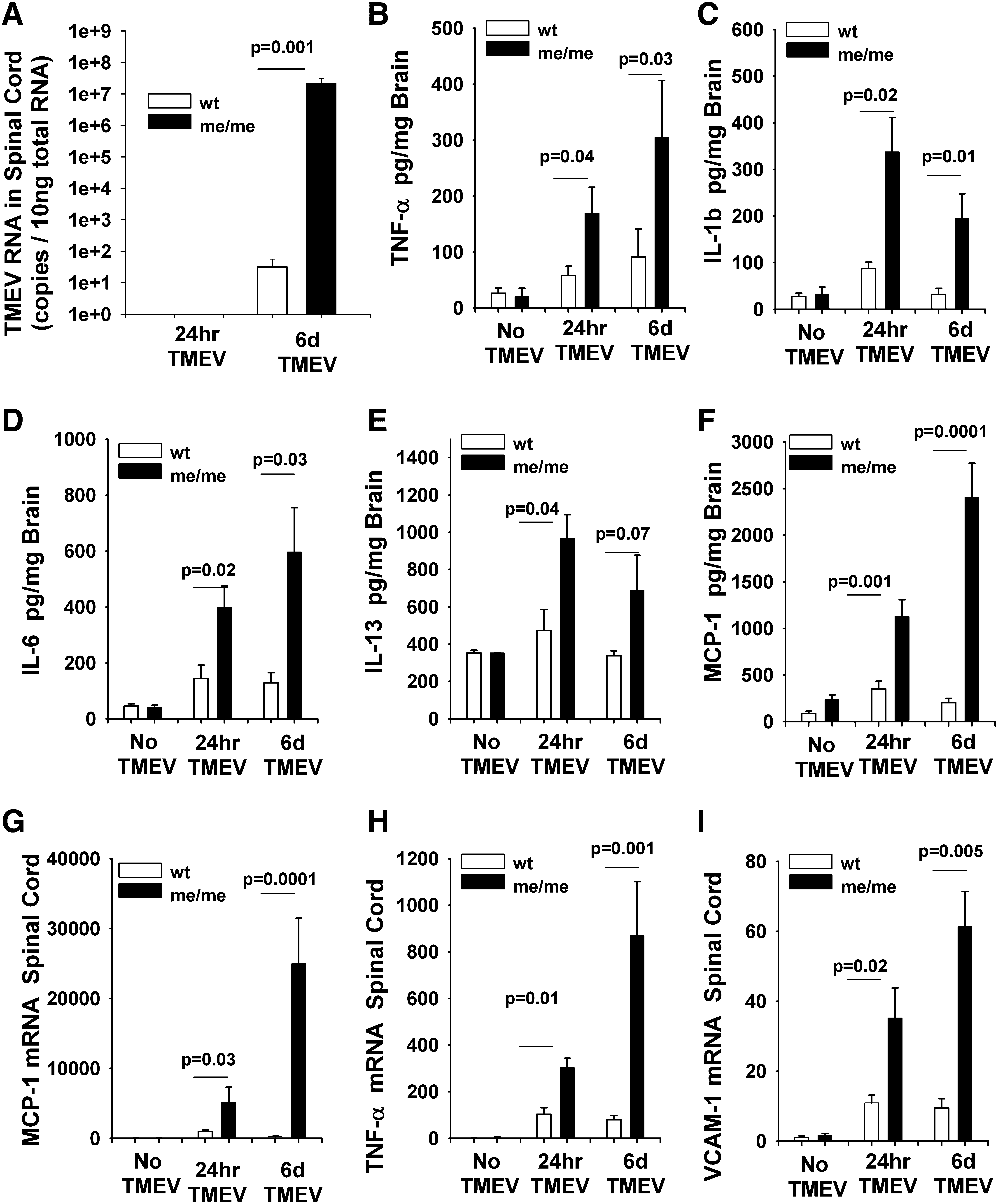
We have shown that IP TMEV infection results in paralysis, macrophage infiltration, and demyelination, exclusively in SHP-1-deficent mice compared to their wild-type littermates. Next, it was important to examine the viral loads and inflammation in the CNS of infected mice at 24 h and 6 dpi to gain insights into both the early and late events in the initiation of inflammatory demyelination. First, the genome levels of TMEV (VP2) were quantified in the spinal cords of wild-type and SHP-1-deficient mice at 1 and 6 dpi after inoculation with 106 PFU BeAn/mouse (Fig. 3A). At 1 dpi no virus was detected in the spinal cords of either wild-type or me/me mice. At 6 dpi the spinal cords of me/me mice had an average of 107 TMEV RNA genome copies, compared to 102 TMEV RNA genome copies seen in wild-type mice. The RNA quantities were corroborated by the levels of TMEV antigens found in spinal cords as analyzed by immunohistochemistry (data not shown).
(
We also measured the levels of several cytokines and chemokines by ELISA in the CNS of wild-type and SHP-1-deficient mice at 24 h and 6 d post-IP inoculation with 106 PFU BeAn/mouse (Fig. 3B–F). Interestingly, at 24 h dpi although no infiltrating cells or virus were detected in the CNS, there was a significant increase in the levels of TNF-α, IL-1β, IL-6, IL-13, and MCP-1 in the brains of me/me mice compared to wild-type mice. Similar results were seen at 6 dpi. The levels of cytokines and chemokines expressed in the CNS correlated well with mRNA levels as represented by both TNF-α and MCP-1 (Fig. 3G and H). In addition, there were increased expression levels of the adhesion molecule VCAM-1 in the brains of me/me mice following IP TMEV infection, indicating that vascular endothelial cells may be similarly affected (Fig. 3I). These data suggest that peripheral inoculation with TMEV may generate an early signal that stimulates production of chemokines and adhesion molecules within the CNS prior to invasion of macrophages and virus into the CNS.
SHP-1-deficient mice have augmented serum cytokine levels following systemic TMEV infection
To implicate a possible peripheral virus-induced stimulus of cytokine, chemokine, and adhesion molecule expression in the CNS of peripherally-inoculated animals at 1 dpi, we analyzed likely candidate cytokines in the blood following peripheral TMEV inoculation (Fig. 4). The levels of TNF-α, IL-1β, IL-6, IFN-γ, IL-13, and MCP-1 were quantified by ELISA of serum of wild-type and SHP-1-deficient mice both before inoculation and at 24 h post-IP-inoculation with 106 PFU BeAn/mouse. Similarly to our observations in the CNS, the serum levels of TNF-α, IL-1β, IL-6, and IL-13 were increased in me/me mice compared to wild-type mice, with the exception of IFN-γ. Similarly, the chemokine MCP-1 was also increased in the serum of me/me mice compared to wild-type mice (Fig. 4F). Based on these data, we reasoned that this early peripheral cytokine/chemokine response may constitute the initial stimulus to resident CNS cells for increased central expression of chemokine/adhesion molecules that increase trafficking of blood-borne monocytes to the CNS.
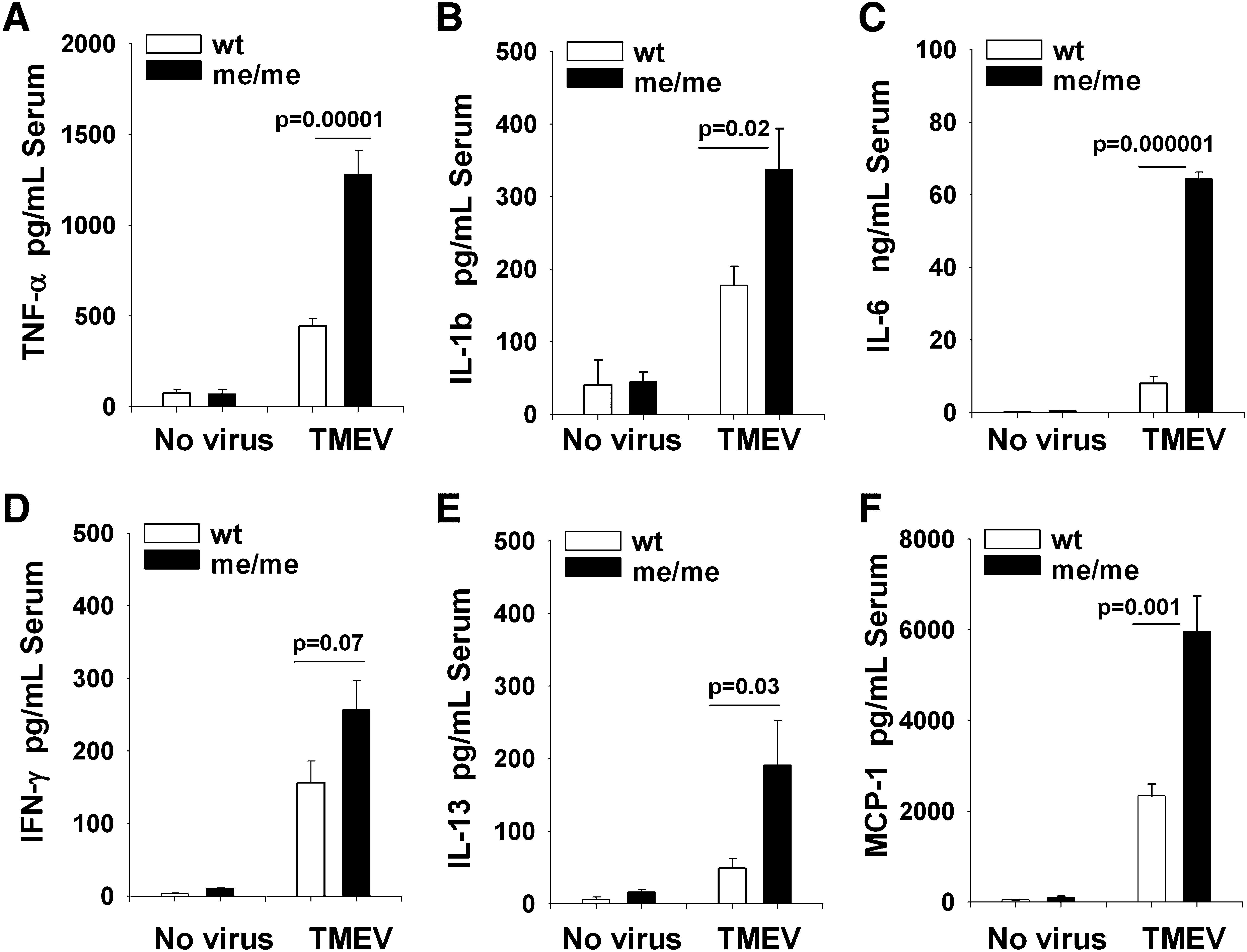
The levels of TNF-α, IL-1β, IL-6, IFN-γ, IL-13, and MCP-1 were quantified in the serum of wild-type (wt) and SHP-1-deficient mice (me) either before (no virus) or mice at 24 h post-IP-infection (TMEV) with 106 PFU BeAn/mouse. ELISA was used to measure the cytokine levels in the serum of wt (n = 8), me (n = 8), wt-24 h post-infection (n = 10), and me-24 h post-infection (n = 8) mice.
Depletion of CNS-infiltrating macrophages inhibits disease development
The above observations implicate a role for infiltrating macrophages in the paralysis, demyelination, and increased viral loads seen in TMEV-infected me/me mice compared to wild-type mice. To directly demonstrate this role, macrophages were experimentally depleted in vivo by injecting mice with liposome-encapsulated clodronate. Clodronate liposomes cause transient and selective elimination of macrophages in the spleen, multiple peripheral tissues, and CD11b+/Ly6Chi monocytes in the blood (39,82). In contrast, clodronate liposomes do not deplete microglia in the brain (5,70). In previous studies by our group and others, clodronate liposomes were successfully used to deplete blood-derived macrophages, thus preventing them from participating as effectors in CNS inflammatory disease (18,33,40,87). In the present experiment, wild-type and me/me mice received either clodronate liposomes or PBS control liposomes as previously described (18), at 2 d before and 2 and 6 d after IP injection of TMEV. Clodronate liposome treatment effectively depleted macrophages (CD45hiCD11b+ cells) from the spleens of both wild-type and me/me mice, but there was no depletion seen with PBS control liposomes (data not shown). Infected me/me mice that were pretreated with clodronate liposomes had significantly delayed signs of paralysis compared to mice treated with control liposomes (Fig. 5A). Furthermore, peripheral macrophage depletion resulted in significantly decreased mortality (64%) in infected me/me mice compared to control-liposome-treated mice (100%). In addition, to assess the effect of macrophage depletion on demyelination in the spinal cords of TMEV-infected me/me mice following treatment with clodronate liposomes, double-color immunohistochemistry for MBP and CD11b was performed (Fig. 6A–D). Spinal cords of TMEV-infected wild-type mice that were treated with either control or clodronate liposomes were free of CD11b+ macrophages and showed no signs of demyelination (Fig. 6A and B). In contrast, spinal cords of infected SHP-1-deficient me/me mice that were treated with control liposomes showed substantial white matter macrophage infiltration associated with extensive demyelination (Fig. 6C). Importantly, macrophage depletion in infected me/me mice resulted in both a disappearance of CD11b+ macrophages and focal white matter demyelination in stained sections of the spinal cord (Fig. 6D). Similar findings of decreased CD11b+ macrophages in spinal cords were obtained with flow cytometry (data not shown). These data show that peripherally-derived CD11b+ macrophages were essential for effecting demyelination in these mice following peripheral inoculation with TMEV.
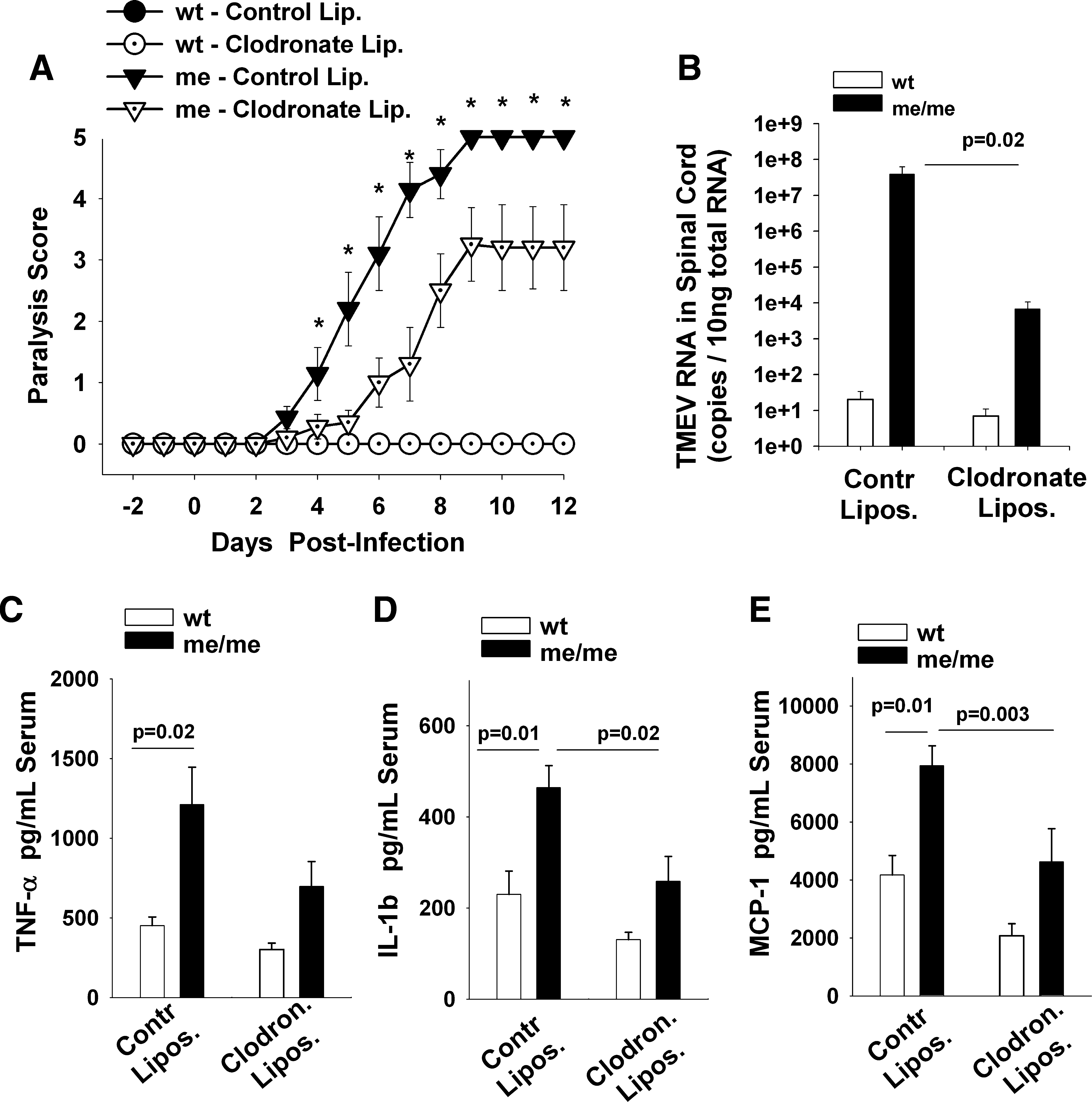
(

Immunohistochemical analysis of MBP, CD11b, and TMEV antigen in spinal cord sections of infected wt and me/me mice that received either control liposomes or clodronate liposomes 2 d before IP infection with 106 pfu BeAn/mouse. At least four mice from each group were examined and the spinal cords were surveyed at different levels. Mice were anesthetized and perfused with paraformaldehyde. Frozen spinal cords were sectioned 8 μm thick. (
Infiltrating macrophages contribute to TMEV neuroinvasion and increased CNS viral loads
Peripheral and CNS-infiltrating macrophages are known targets for TMEV infection, and significantly contribute to both virus burden and the process of inflammatory demyelination in spinal cords of mice (23,54,59,64). Because TMEV infection of the CNS in the experiments described here originated in the periphery, the main targets of TMEV infection in mice are macrophages, and macrophages are common conduits of virus neuroinvasion; thus we assessed the importance of peripheral macrophages in TMEV neuroinvasion and virus burden in me/me mice following IP infection. The amount of viral RNA (Fig. 5B) and viral antigen (Fig. 6E–H) were examined at 6 dpi in the spinal cords of TMEV-infected mice that had been pretreated with either control or clodronate liposomes. Viral RNA was significantly reduced in the spinal cords of clodronate-liposome-treated compared to control-liposome-treated infected me/me mice, from 108 copies down to 104 copies (Fig. 5B). Similarly, the presence of the TMEV antigen in the spinal cords of infected mice was measured with immunohistochemistry (Fig. 6E–H). Spinal cords of TMEV-infected wild-type mice that were either treated with control or clodronate liposomes did not show any signs of TMEV antigen (Fig. 6E and F). In contrast, spinal cords of infected SHP-1-deficient me/me mice that were treated with control liposomes showed substantial TMEV antigen throughout the spinal cord, in the gray and white matter, and particularly in infiltrating macrophages (Fig. 6G). Importantly, macrophage depletion in infected me/me mice with clodronate liposomes resulted in a substantial decrease in TMEV antigens in spinal cord sections (Fig. 6H). These data suggest that SHP-1 may control virus neuroinvasion via macrophages in the CNS.
SHP-1-deficient macrophages are more susceptible to viral infection and have a pronounced inflammatory profile
Because macrophages were the major infiltrating leukocyte seen in the CNS of me/me mice, and macrophage depletion resulted in a dramatic decrease in CNS viral loads following systemic TMEV infection, we asked whether increased replication rates in SHP-1-deficient macrophages could be additionally responsible for the increased CNS virus burden in me/me mouse cords. Therefore, cultured macrophages from wild-type and me/me mice were infected with TMEV for 48 h and the viral genome levels, virus antigens, and inflammatory profile were assessed (Fig. 7). SHP-1-deficient macrophages had significantly higher TMEV RNA and antigen compared to wild-type macrophages (Fig. 7A and B). Furthermore, TMEV infection resulted in increased secretion of the cytokines TNF-α, IL-1β, and IL-6, and MCP-1 in the supernatants of SHP-1-deficient macrophages compared to wild-type macrophages. In addition, the protease ADAM8, which plays an important role in both trans-endothelial migration and proteolysis of myelin proteins by macrophages in inflammatory demyelinating diseases (2,72,93), was also significantly higher in me/me macrophages compared to wild-type macrophages (Fig. 7H). These data indicate that a given inoculation of either wild-type or me/me macrophages produces a greater replication rate of TMEV and generation of inflammatory molecules in SHP-1-deficient macrophages, which is likely to increase the demyelinating activity of these cells as they migrate into the CNS.
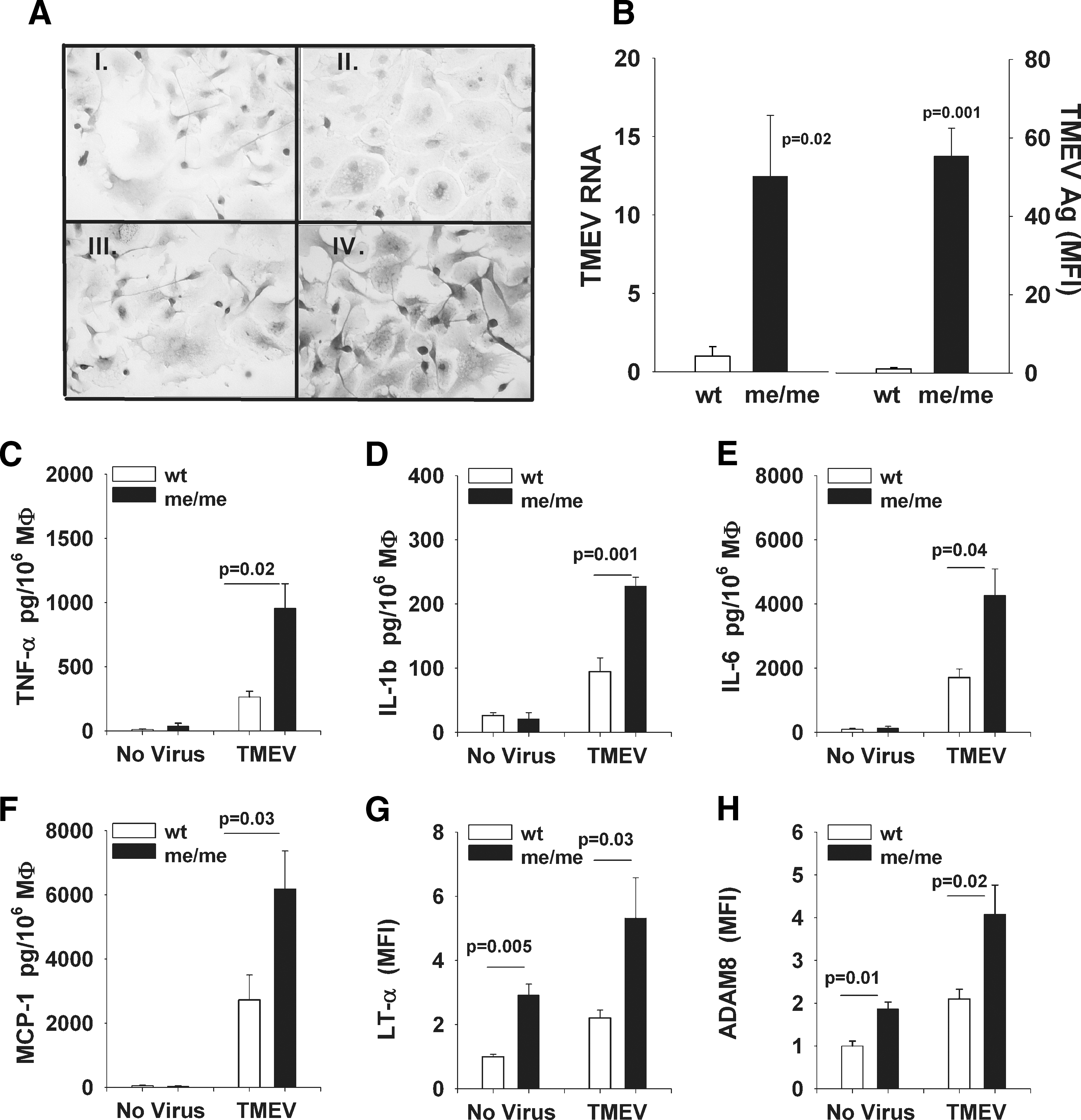
Splenic macrophages from SHP-1-deficient mice are more susceptible to TMEV infection and have an exacerbated inflammatory profile following TMEV infection. Splenic macrophages were expanded in the presence of CSF-1, and were infected with BeAn TMEV at an MOI of 1 for 48 h. (
MCP-1 is an important mediator of TMEV-induced disease
MCP-1 has repeatedly been shown to play a central role in chemoattracting macrophages and modulating the severity of disease both in the TMEV (6,44,56) and EAE (4,42) models of MS. We have previously shown that MCP-1 may play an essential role in accumulation of macrophages and demyelination in the CNS of SHP-1-deficient mice following intracerebral inoculation of TMEV. These studies showed that TMEV-infected me/me glial cells and the CNS of TMEV-infected me/me mice produced increased amounts of the NF-κB-inducible chemokine MCP-1 (18) (Fig. 3). Furthermore, we demonstrated that SHP-1-deficient macrophages displayed enhanced chemotaxis towards MCP-1 compared to wild-type macrophages (18). To analyze the role of MCP-1 in the present studies, me/me mice were treated with an MCP-1-neutralizing antibody or a control IgG antibody at the time of IP TMEV inoculation, and clinical signs were recorded thereafter (Fig. 8A). MCP-1 neutralization resulted in delayed onset of paralysis in infected me/me mice, suggesting that MCP-1 is an important mediator of inflammatory demyelination in SHP-1-deficient mice. MCP-1 neutralization also resulted in decreased numbers of infiltrating macrophages in the CNS of me/me mice following peripheral TMEV inoculation (Fig. 8B)
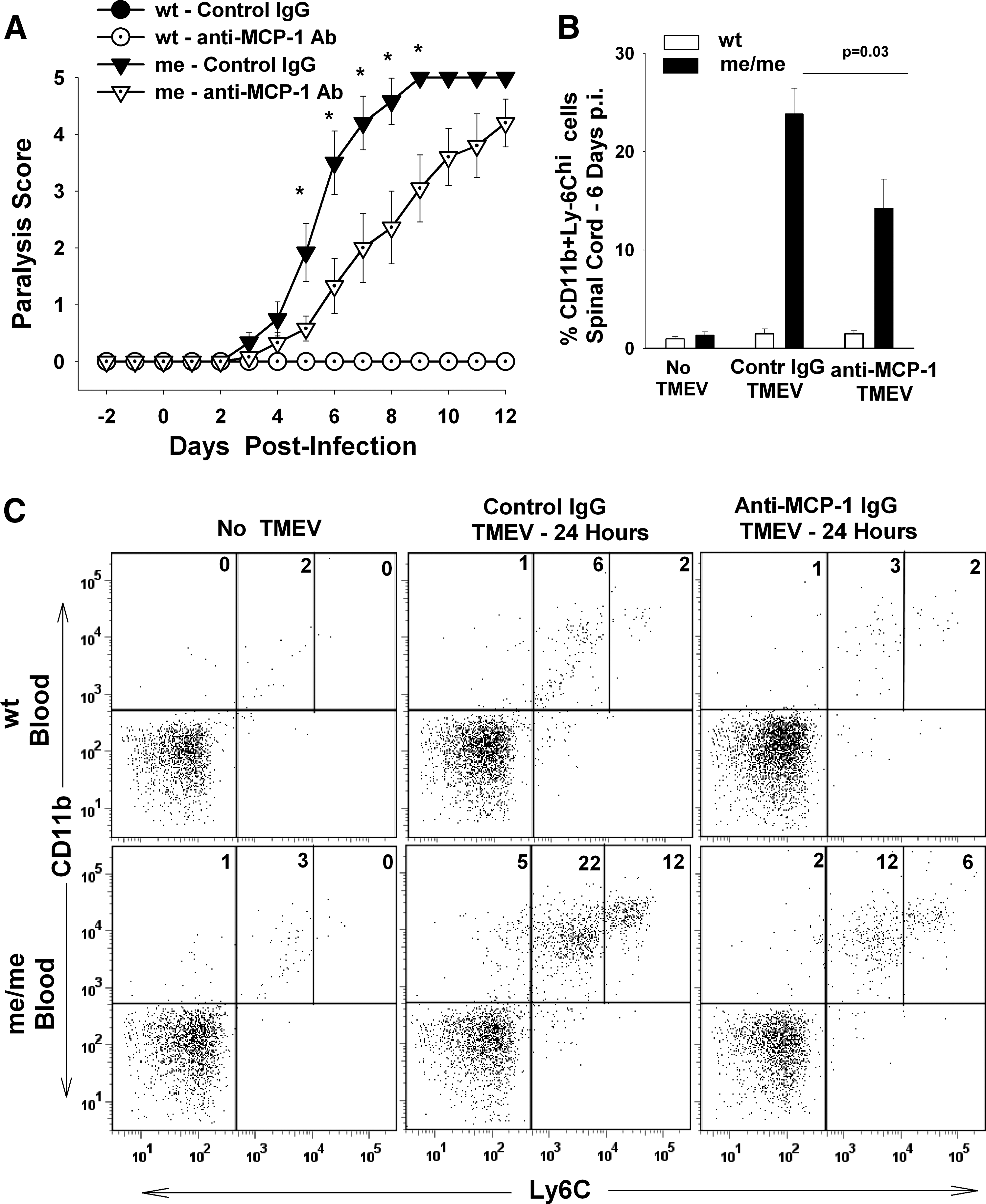
The role of MCP-1 in paralysis, CNS macrophage infiltration, and macrophage egress from bone marrow following systemic TMEV infection. Mice received 60 μg of either control IgG or anti-MCP-1 antibodies on the day of IP infection with 106 pfu BeAn/mouse. Six groups of mice were used in this experiment: wt not infected (n = 10), me not infected (n = 8), wt control IgG (wt - control IgG; n = 8), wt anti-MCP-1 Ab (wt - anti-MCP-1 Ab; n = 5), me control IgG (me - control IgG; n = 10), and me anti-MCP-1 Ab (me - anti-MCP-1 Ab; n = 9). (
MCP-1 neutralization blocks an increase in peripheral blood-borne monocytes following peripheral TMEV infection
During CNS virus infections, CNS-derived MCP-1 is responsible for stimulating macrophage trafficking into the CNS though the formation of a concentration gradient across the blood–brain barrier (BBB), thus attracting blood-borne monocytes through the BBB into the CNS. Additionally, recent reports suggest that peripheral MCP-1 (as shown Fig. 5E) also functions to promote monocyte egress from the bone marrow to the blood, thus effectively increasing the number of circulating monocytes available to migrate to inflammatory sites (41,58,89). Therefore we quantified the number of circulating blood monocytes in TMEV-infected mice treated with control IgG or anti-MCP-1 IgG (Fig. 8C). As expected, before infection wild-type and me/me mice had only 2–4% of circulating monocytes out of the entire leukocyte population in the blood. However, following IP TMEV inoculation, circulating monocytes in control IgG-treated me/me mice increased to 39% of all leukocytes in the blood, compared to 9% seen in wild-type mice (Fig. 8C). Importantly, MCP-1 neutralization decreased the number of circulating monocytes by half, both in infected wild-type and me/me mice (Fig. 8C). Taken together, these data suggest that in addition to attracting macrophages into the CNS, MCP-1 is critical for stimulating increased numbers of monocytes in the blood immediately following TMEV infection, and therefore may constitute a critical component in increased infiltration of macrophages into the CNS of me/me mice.
Discussion
We have previously shown that intracranial TMEV infection results in an acute macrophage-mediated inflammatory demyelination in SHP-1-deficient mice (18,60). The present study was aimed at investigating the role of SHP-1 in mediating inflammatory demyelination following peripheral TMEV inoculation. Herein we show that IP TMEV inoculation results in CNS macrophage infiltration, virus neuroinvasion, and macrophage-mediated inflammatory demyelination, exclusively in SHP-1-deficient mice compared to their wild-type littermates. In turn, macrophage depletion or MCP-1 neutralization resulted in a significant delay in the onset and severity of paralysis, suggesting that peripheral macrophage activation and infiltration into the CNS is essential for the development of demyelination.
TMEV antigen (27), virions (9), and viral RNA (3) have been detected in macrophages in demyelinating lesions in mice, indicating that macrophages promote disease progression by providing an essential source of virus replication and innate inflammatory signals in the CNS (23,52,54,59,64). This study demonstrates that macrophages are essential in mediating the virus neuroinvasion into the CNS, and provide viral targets for TMEV to sustain productive CNS infection. We have previously shown that SHP-1 deficiency in CNS glia results in higher viral replication both in vivo and in vitro, correlating with decreased nitric oxide production (11,60). Furthermore, virus infection of me/me macrophages induces expression of multiple proinflammatory molecules by these cells. Taken together, SHP-1 controls both the number of macrophages entering the CNS and viral replication within macrophages, which in turn both contribute to the increased virus and inflammation seen in the spinal cords of me/me mice.
We have shown that in SHP-1-deficient mice, following systemic TMEV infection, CNS viral entry is macrophage-dependent. The fact that SHP-1 controls viral replication in macrophages and macrophage infiltration into the CNS may have general significance for understanding virus-induced CNS disease, especially where virus entry is macrophage-mediated, such as in HIV, HTLV-1, or West Nile virus infections (47,69,73). Furthermore, the observation that SHP-1 controls viral replication may have important implications in the pathogenesis of MS, since macrophages of MS patients display decreased SHP-1 levels. Therefore it will be important to investigate whether macrophages of MS patients are more susceptible to virus infections, or if they display increased innate activation following viral infection that would promote enhanced migration into the CNS.
There is compelling evidence that formation of white matter lesions in multiple sclerosis is immune mediated (32,66). It is widely thought that the initiation of MS is the result of a systemic infection that causes proinflammatory cytokines to be released into the general circulation, subsequent upregulation of adhesion-related molecules on CNS endothelium, homing of leukocytes to the CNS vasculature, and infiltration of inflammatory leukocytes to CNS lesions. This hypothesis is supported by immunopathological descriptions of the MS brain, and corresponding observations from animal models of MS. Several researchers postulate that a viral infection might trigger MS (53) by several mechanisms in which the virus acts as an adjuvant, provides autoantigenic determinants of molecular mimicry, promotes epitope spreading, or causes persistent CNS infection and inflammation (65). In support of these ideas, viral infection has been closely associated with MS onset by pathways that may be similar to those described for several viruses in MS animal models (68).
One potential limitation of viral mouse models for MS is that these viruses, including TMEV, mouse hepatitis virus (MHV or JHMV), and Visna lentivirus, are by necessity intracerebrally inoculated into susceptible animals in order to develop inflammatory demyelination (35,68,85,86). While useful to study events occurring subsequent to CNS infection, these models do not allow an examination of the steps leading up to demyelinating disease that unfold following virus infection at peripheral sites that may be important in the pathogenesis of MS. Therefore attempts have been made to identify particular viruses or appropriate susceptible mouse strains in which a peripheral viral infection can lead to central inflammatory demyelination. Peripheral TMEV infection of SHP-1-deficient mice as described here may provide such a model by which early peripheral events leading to white matter inflammation may be thoroughly examined.
This study substantiates the idea that systemic viral infection can specifically cause CNS inflammatory demyelinating disease in individuals with a stable deficiency in SHP-1. Based on the important and diverse roles that SHP-1 has on macrophage biology, including controlling (1) the susceptibility to viral infections, (2) the production of inflammatory molecules, (3) the responsiveness to chemoattractive stimuli, and (4) the rate of phagocytosis, SHP-1 deficiency could potentially enhance several key steps leading up to macrophage-mediated demyelination, making SHP-1 an attractive target for the treatment of MS (Fig. 9) (18,38,43,46,67). Indeed we have shown that monocyte-derived macrophages of MS patients have deficient levels of SHP-1 relative to those of normal control subjects (20). Furthermore, we have shown that deficient levels of SHP-1 seen in macrophages in MS patients may be directly responsible for the increased activation of inflammatory transcription factors, as seen in SHP-1-deficient mice. In addition, we have shown that SHP-1 has interferon-β-inducible activity, and that SHP-1 induction following interferon treatment of MS patients attenuates the leukocyte inflammatory profile (17,21). These observations indicate that heightened SHP-1 expression may be a useful biomarker of drug efficacy in MS patients.
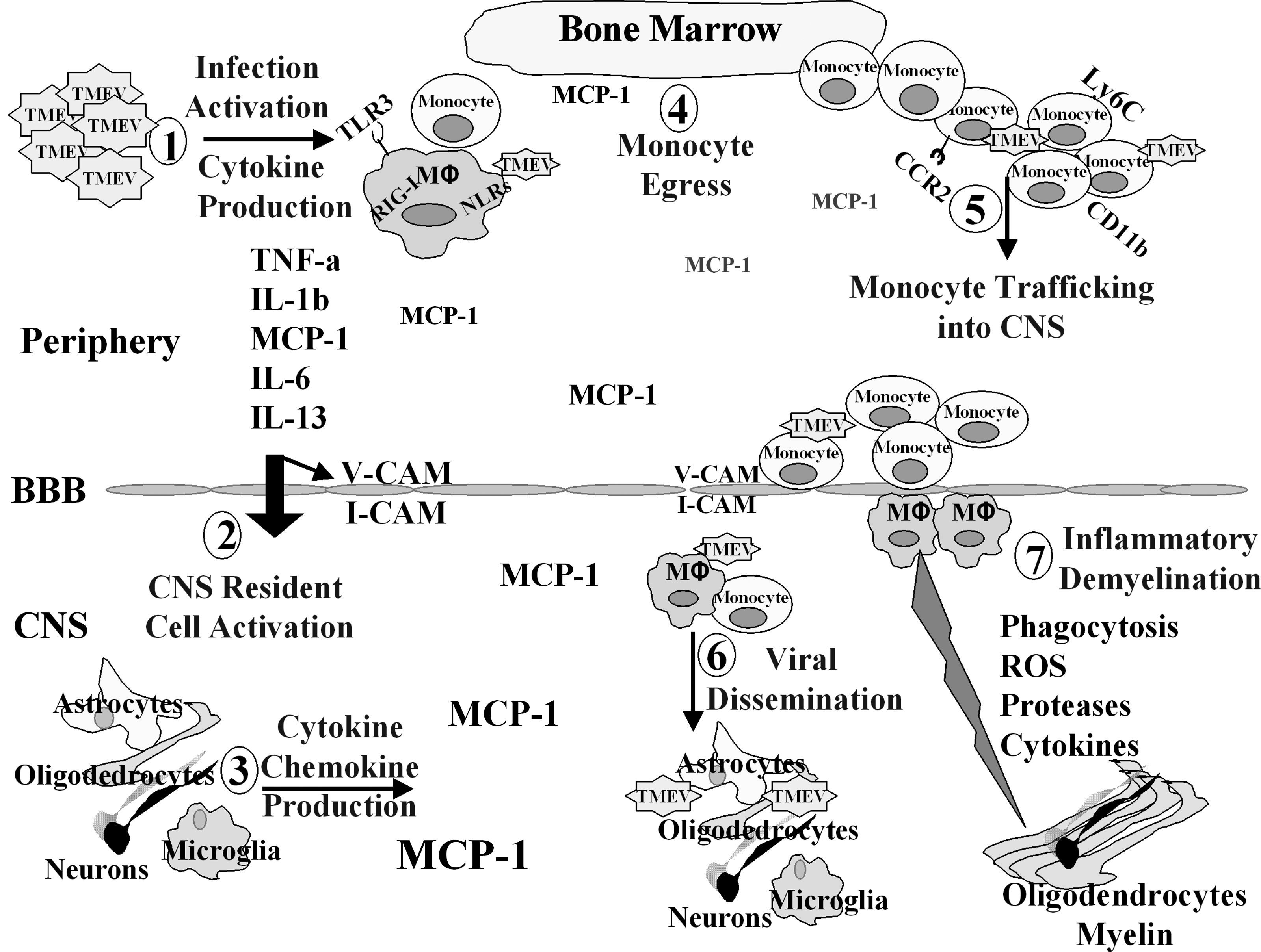
The potential mechanisms of inflammatory demyelination in SHP-1-deficient mice following systemic TMEV infection. (1) Following IP TMEV infection the virus activates immune cells to secrete cytokines and chemokines. Furthermore, TMEV selectively infects me/me macrophages, leading to macrophage activation. This cytokine storm leads to upregulation of several inflammatory molecules, including chemokines, proteases, and adhesion molecules. (2 and 3) Peripheral cytokines can cross the blood–brain barrier (BBB) and enter the CNS. This results in CNS resident cell activation, including microglia, astrocytes, and BBB endothelial cells, leading to the upregulation of several inflammatory mediators, including the chemokine MCP-1. (4) MCP-1 secreted from the CNS and the periphery can mediate monocyte egress from the bone marrow that can substantially increase the number of circulating monocytes. The majority of circulating monocytes are activated, as is evident by the high expression of the Ly6C marker. The fact that me/me mice secrete more MCP-1 following viral infection, and that SHP-1-deficient macrophages are more responsive to chemoattractive stimuli, leads to a substantially higher number of peripheral monocytes/macrophages in me/me mice compared to wt mice. (5) The increased number of circulating monocytes is attracted into the CNS, largely by the chemokine MCP-1. Monocytes entering the CNS introduce TMEV, a neurotropic virus, into the CNS. (6) TMEV disseminates into the CNS, infecting glial cells, neurons, and infiltrating macrophages. This leads to further inflammatory mediators being released into the CNS, and this in turn contributes to additional CNS macrophage infiltration. The CNS-infiltrating macrophages are very susceptible to TMEV infection, thereby contributing to the increased viral loads observed in the CNS of SHP-1-deficient mice. (7) Infiltrating macrophages can directly cause demyelination by myelin phagocytosis. Also, several inflammatory molecules expressed or secreted by infiltrating macrophages, such as reactive oxygen species (ROS), proteases, or cytokines, can contribute to myelin degradation and oligodendrogliopathy mθ.
In MS, intense macrophage infiltration is present in active demyelinating lesions, and both macrophage numbers and differentiation/activation correlate with disease severity (15,16,55,88,91). Macrophages can mediate myelin degradation, oligodendrogliopathy, and axonal loss, both through cell-mediated processes and the secretion of inflammatory mediators (37,66,91). A key area of interest is the stimulus responsible for macrophage trafficking/infiltration into CNS demyelinating lesions. For instance, the chemokine MCP-1 is elevated in MS plaques and in the cerebrospinal fluid of MS patients (56,63,81). An essential role for MCP-1 in infiltration of macrophages in CNS demyelinating diseases is supported by observations in various animal models for MS. These findings point out that CNS-derived chemokines and peripherally-derived macrophages have a prominent role in the initiation and progression of demyelination in MS. However, the exact interplay of MCP-1, MCP-1-producing cells, and macrophages in MS and in relevant animal models needs to be further studied.
In the present study, we considered the specific mechanisms of increased macrophage recruitment into the CNS of me/me mice following peripheral TMEV infection. It was previously shown that the CNS of the uninfected me/me mouse possess an intact BBB, and that the CNS contains normal numbers of monocytic cells (92). Previous studies have shown that in response to inflammatory stimuli, macrophages of me/me mice secrete increased levels of chemokines compared to wild-type animals (31). Thus MCP-1 produced by infected macrophages may constitute an initial stimulus to promote increased monocyte egress from the bone marrow into the blood of infected me/me mice in the present study, and as shown in previous studies (41,58,89). Also, our previous studies showing that blocking MCP-1 in vitro significantly diminished macrophage migration in response to glial-derived MCP-1 indicates an additional role of MCP-1 in attracting blood-borne monocytes into the CNS (18).
A recent study analyzed a novel mouse strain with SHP-1 deficiency (the so-called spin mouse) in which SHP-1 deficiency was determined necessary but not sufficient to cause systemic inflammation and autoimmunity. Indeed, spin mice derived under sterile conditions and housed in a germ-free environment were entirely healthy until transferred to a nonsterile environment, in which these mice developed spontaneous inflammatory lesions in skin and lungs commonly seen in me/me mice (24). These observations indicate a two-step process in which SHP-1 deficiency in macrophages is a precondition to tissue-specific inflammation triggered by microbe-stimulated inflammatory signals. This scenario is in agreement with the present study, demonstrating that upon introduction of a neurotropic virus either directly into the CNS or at peripheral sites, inflammation via innate pathways ensues in the CNS. Thus a remaining question is the extent to which loss of SHP-1 in either peripheral macrophages or CNS-resident cells contributes to microbe-induced inflammation in the CNS. Our previous studies of direct CNS infection may indicate a role for dysregulation of innate responses to virus infection within CNS-resident cells (11,60). The answer to this important question awaits studies in which the role of SHP-1 in either peripheral myeloid or CNS-resident glia may be studied individually using targeted ablation in these populations in TMEV-infected mice.
Footnotes
Acknowledgments
This work was supported by research grants from the National Multiple Sclerosis Society (RG2569C5) and NIH grant (NS041593) to Paul T. Massa. We would like to thank Dr. Isobel Scarisbrick at Mayo Clinic for providing the cDNA plamids used in performing real-time RT-PCR, and Howard Lipton for providing the anti-TMEV antibody. We would also like to thank Dr. Nick J. Gonchoroff, for his expertise in the flow cytometry/cell sorting performed in this study.
Author Disclosure Statement
No conflicting financial interests exist.