
Abstract
Two major nuclear factor-κB (NF-κB) signalling pathways are involved in the regulation of the immune response. While the classical NF-κB pathway is responsible for regulation of genes encoding components of the innate immune response, the alternative NF-κB signalling pathway mediates processes of the adaptive immune system. To evaluate the role of the NF-κB signalling pathways in the control of viral infection, we have used lymphocytic choriomeningitis virus (LCMV) infection of mice, which is known to be an excellent model for studying antiviral immune responses. Via the use of mice that were deficient in NF-κB subunits from either the classical (p50−/− mice) or the alternative NF-κB pathway (p52−/− mice), we were able to demonstrate that the alternative NF-κB pathway is required for the T-cell-mediated immune response against LCMV. Mice that were deficient in the alternative NF-κB pathway subunit p52 showed an impaired T-cell response against LCMV infection. Furthermore, these mice also showed an impaired T-cell-dependent humoral immune response against vesicular stomatitis virus (VSV) infection. Adoptive transfer experiments revealed that impaired priming, but not the T-cell response itself, was responsible for the defective cellular immune response against LCMV infection. Our data demonstrate that a functional alternative NF-κB signalling pathway is required to assure an adequate immune response after viral infection.
Introduction
The immune response seen after infection with the non-cytopathic lymphocytic choriomeningitis virus (LCMV) is well characterized (54,72). The course of LCMV infection in mice depends on the virus dose, infection route, and the age of the infected mouse. In immunocompetent mice an acute LCMV infection is efficiently controlled by cytotoxic effector mechanisms, namely natural killer (NK) cells and cytotoxic T-lymphocytes (CTL) (20,27,33,53,60,73). Virus elimination during acute phase infection mainly depends on the massive induction of virus-specific CD8+ T cells. The maximum CTL activity is seen by day 8 after LCMV infection (10,21,22). Transplacental or neonatal infection of mice or infection of immunodeficient mice leads to a lifelong, persistent infection (9,31,53,55,56). Intracerebral inoculation of LCMV generally results in fatal T-cell-mediated meningitis (15,24,47,48). LCMV infection of mice serves as an excellent model to study the immunological mechanisms that are required for defense against viral infections. Although our knowledge of the mechanisms of the cellular immune response against viral infection has increased tremendously in recent years, the influence of NF-κB on this process is not very well understood. The knowledge of how NF-κB is involved in the immune defense against viral pathogens, however, could help to improve therapeutic strategies against viral infections. Thus the present study was focused on better understanding of the role of NF-κB in immunity against viral infection. We demonstrate that the alternative NF-κB pathway is a prerequisite for the T-cell-mediated immune response against LCMV. Mice that were deficient in the NF-κB subunit p52 showed an impaired T-cell response against this virus. Interestingly, T cells from NF-κB p52−/− mice were responsive when transferred into wild-type mice, indicating the importance of the alternative NF-κB pathway for priming rather than for signalling processes in the T cell itself.
Materials and Methods
Virus
LCMV, WE-strain, was obtained from R.M. Zinkernagel (Institute of Experimental Immunology, Zürich, Switzerland) and further propagated on L929 cells at a low multiplicity of infection (MOI) at the Friedrich-Loeffler-Institute, Federal Research Institute for Animal Health, Tübingen, Germany. Vesicular stomatitis virus (VSV) Indiana was also obtained from R.M. Zinkernagel.
Animals and infections
C57BL/6 mice were obtained from the animal breeding facilities at the Friedrich-Loeffler-Institute, Federal Research Institute for Animal Health, Tübingen, Germany. NF-κB2 (p52−/−) mice were originally obtained from Schmidt and Paxian from the Department of Internal Medicine II, Technical University of Munich. The generation of NF-κB2−/− mice has been described elsewhere (59). NF-κB2−/− mice were back-crossed into the C57BL/6 strain at least 10 times at the animal breeding facilities of the Friedrich-Loeffler-Institute. NF-κB1 (p50−/−) mice were purchased from Jackson Laboratories (Bar Harbor, ME) and were back-crossed onto a C57BL/6 background as previously described (8,16).
Female mice at the age of 6–8 wk were used throughout all the experiments. For LCMV infection, mice were either infected with 1 × 102 plaque-forming units (pfu) or 1 × 106 pfu injected intravenously into the tail vein, or were infected intracerebrally with 1 × 103 pfu. For VSV infection, the animals were infected with 2 × 106 pfu VSV-IND injected intravenously into the tail vein.
Cells
The murine fibroblast line MC57 obtained from C57BL/6 mice (H-2b) was used for in-vitro infections with LCMV-WE. The cells were cultured with minimum essential medium (MEM; GibcoBRL, Carlsbad, CA) supplemented with 5% FCS (PAA Laboratories, Pasching, Austria) and 2 mM L-glutamine and 100 U/mL gentamicin.
Infectivity assay
To assess the number of infectious particles (plaque titers) in blood and organs of LCMV-infected wild-type NF-κB p50−/− and p52−/− mice, standard focus-forming assay on MC57 cells (5) was performed in 24-well plates. Virus-infected cells were immunostained by incubating for 1 h with a monoclonal antibody specific for the LCMV nucleoprotein (VL4), followed by a 30-min incubation with an anti-rat biotin-labeled antibody (Dianova, Hamburg, Germany), and by a 30-min incubation with streptavidin-peroxidase conjugate (Dianova). The reaction was visualized with ortho-phenylenediamine substrate (Sigma-Aldrich, St. Louis, MO). Titers are expressed as pfu per milliliter of 10% organ homogenates or per milliliter of blood. The detection limit of the focus-forming assay was 1.7 log10 pfu/mL.
Footpad swelling reaction
Delayed-type hypersensitivity (DTH) response was induced by injecting 1.5 × 103 pfu LCMV-WE into the left hind footpad of either four C57BL/6 NF-κB p50−/− or NF-κB p52−/− mice. Footpad thickness was measured with a spring-loaded caliper (Kroepelin, Schluchtern, Germany). Increased footpad thickness was expressed as the percent swelling relative to the thickness of the uninfected right hind footpad.
T-cell depletion
For the depletion of T cells, C57BL/6 NF-κB p52−/− mice were inoculated intraperitoneally (IP) with monoclonal antibodies against CD4 (YTS 191.1, rat IgG2b) or CD8 (YTS 169.4, rat IgG2b) 2 wk prior to the experiment. Antibodies were produced as ascitic fluids and purified by affinity chromatography. For depletion of CD4 cells mice were treated with the anti-CD4-specific antibody YTS 191.1 diluted 1:500 in balanced salt solution (BSS). CD8 cells were depleted by injection of the anti-CD8-specific antibody YTS 169.4 diluted 1:200 in BSS at 4-day intervals. The depletion of T lymphocytes was assessed using FACS analysis with monoclonal antibodies to CD8a (clone 53-6.7), CD4 (clone H129.19), and CD3e (clone145-2c11, all from BD Biosciences Pharmingen, Franklin Lakes, NJ). Only mice with complete depletion of T cells were used for further experiments.
Cytotoxic T-cell response
Spleens from C57BL/6 NF-κB p52−/− mice were harvested on day 8 post-IV-LCMV infection with 1.5 × 102 pfu. For the assay of CTL activity, MC57 target cells were either infected with LCMV-WE at a MOI of 0.1 or were left uninfected. Two days later the cells were labeled with Na2 51CrO4 and used as targets in a 6-h 51Cr-release assay (57). Single-cell suspensions of spleen cells were used as effector cells at the indicated effector:target ratios. Cytotoxicity assays were performed in MEM supplemented with 2% FCS, 2 mM L-glutamine, and 100 U/mL gentamicin. At the end of the 6-h incubation period, the plates were centrifuged and 25 μL of supernatant from each well was harvested and counted in a Wallac microbeta counter (PerkinElmer, Waltham, MA). For determining spontaneous and maximum release, target cells were only incubated with medium without effector cells or 1 N HCl, respectively.
The percentage of specific lysis was calculated as (experimental release − spontaneous release)/(total release − spontaneous release) × 100. Spontaneous release was always <20%.
Measurement of VSV-specific antibody responses
To determine total Ig or IgG serum titers against VSV, mice were infected by IV injection into the tail vein with 2 × 106 pfu VSV-IND. On days 0, 4, 8, and 12 post-infection (p.i.) the animals were bled and serum samples were collected. Polystyrene microtiter plates were coated and kept at 4°C overnight with 0.5 μg of purified VSV per 100 μL of carbonate-bicarbonate buffer (pH 9.6) per well. The plates were washed five times with phosphate-buffered saline (pH 7.2) containing 0.05% Tween-20, and then 100 μL of serial dilutions of sera in phosphate-buffered saline plus 0.05% Tween 20 was added to each well and incubated for 2 h at room temperature. The plates were washed again, and 100 μL of goat anti-mouse total Ig or goat anti-mouse IgG labeled with horseradish peroxidase (HRP) diluted optimally in phosphate buffered saline plus 0.05% Tween 20 was added, and then the plates were incubated for 2 h at room temperature. The plates were washed again, and 200 μL of substrate containing 2 mg of 2,2′-azino-di-3-ethylbenzthiazoline sulfonate in 20 mL of 0.1 M NaH2PO4 (pH 4.0), with 15 μL of 30% H2O2 was added to each well. After 30 min of incubation at room temperature, all samples and the appropriate controls were measured at 405 nm with a micro-ELISA reader.
Detection of anti-LCMV antibodies
For detection of the different LCMV-specific Ig subtypes (IgM and IgG) and the IgG subclasses, an enzyme-linked immunosorbent assay (ELISA) was performed. Serum samples from high-dose (106 pfu) LCMV-WE infected mice were used. ELISA plates were coated overnight at 4°C with baculovirus-derived LCMV-NP (1 μg/mL) in carbonate buffer (pH 9.6). Afterwards the plates were blocked with 1% BSA-PBS/0.05% Tween-20, 40-fold–prediluted sera in 1% BSA-PBS/0.05% Tween-20, titrated 1:2 over eight dilution steps were added. After washing, the plates were incubated with the HRP-conjugated secondary antibodies anti-mouse IgM, IgG, IgG1, IgG2a, IgG2b, or IgG3 (Southern Biotechnology Associates, Birmingham, AL). Incubations were performed at room temperature for 60 min; PBS/0.05% Tween-20 was used for all washing steps. Color reactions were performed at room temperature for 20 min with 2.2′-azino-bis-[3-ethylbenzthiazoline-6-sulfonate] (Boehringer Mannheim, Mannheim, Germany) substrate and H2O2. Optical density (OD) was measured at 405 nm by an ELISA reader, and antibody titers were determined as the highest dilution of serum yielding an OD of twice background levels (naive mouse serum) or higher.
Immunohistochemistry
Mice were infected IV with 1 × 106 pfu LCMV-WE. At the indicated time points freshly removed organs were immersed in tissue freezing medium and flash-frozen in liquid nitrogen. Then 7-μm sections were cut in a cryostat, placed on slides, air dried, fixed with acetone for 10 min, and stored at −80°C. For blocking of endogenous peroxidase activity rehydrated sections were incubated for 10 min with 0.3% H2O2 in methanol. After blockage of unspecific binding with 5% goat serum for 30 min, the sections were incubated with the primary antibody to CD4 (BD Biosciences Pharmingen) or CD8a (BD Biosciences Pharmingen) diluted (1:50 and 1:10, respectively) in PBS/5% goat serum overnight at 4°C. Subsequently, after washing of the sections with PBS (three times for 5 min each) they were incubated with the secondary biotinylated goat anti-rat IgG antibody (1:1000; Dianova) for 30 min at room temperature. Afterward incubation with streptavidin-avidin-biotin peroxidase complex for 30 min at room temperature (ABC Peroxidase Elite; Vector Laboratories, Burlingame, CA) was followed by visualization of the peroxidase activity by DAB. The counter-staining was performed with hematoxylin.
Flow cytometry
The mice were infected by IV injection into the tail vein with a high dose of LCMV-WE (1 × 106 pfu/100 μL), and at the indicated time points the animals were bled, and the spleens were removed and mechanically disrupted by passage through a 70-μm cell strainer. A single-cell suspension was stained in FACS buffer (PBS, 2% FCS, and EDTA, pH 7.5) with a 1:100 dilution of the required fluorochrome conjugated monoclonal antibodies to CD8a (clone 53-6.7), CD4 (clone H129.19), and CD3e (clone145-2c11, all from BD Biosciences Pharmingen) for 30 min at 4°C. Before FACS analysis, the cells were washed, erythrocytes were lysed, and the cells were fixed with FACS lysing solution (BD Biosciences Pharmingen). Flow cytometry was performed on a dual-laser FACS-Calibur device and analyzed with Cell Quest software (BD Biosciences Pharmingen).
Adoptive transfer of spleen cells
Donor NF-κB p52−/− and wild-type mice were infected IV with 102 pfu LCMV-WE. At 8 or 60 days post-infection single-cell suspensions of spleens were prepared by passage through a 70-μm cell strainer. Isolated spleen cells were washed twice, counted, and 5 × 107 donor cells were injected IV in a volume of 500 μL BSS into the tail vein of either NF-κB p52−/− or wild-type thymectomized recipients. Recipient mice were thymectomized at the age of 4 wk and additionally treated with monoclonal antibodies against CD4 (YTS 191.1, rat IgG2b) or CD8 (YTS 169.4, rat IgG2b) 2 wk prior to the experiment to remove residual T cells. Successful depletion of T lymphocytes was assessed using FACS analysis with monoclonal antibodies to CD8a (clone 53-6.7), CD4 (clone H129.19), and CD3e (clone145-2c11, all from BD Biosciences Pharmingen) as described above.
Determination of mouse CCL21
The amount of CCL21 in the spleens of the mice was assessed using the DuoSet® Mouse CCL21 (6 Ckine) ELISA kit (R&D Systems, Inc., Minneapolis, MN). The animals were infected IV with LCMV-WE (106 pfu), and at the indicated time points spleens from sacrificed mice were homogenized to a 10% homogenate in saline buffer. The supernatants of the 10% spleen homogenates were used in the CCL21 ELISA according to the manufacturer's protocol.
Statistical analysis
Error bars are given as the standard error of the mean. The Student's t-test was performed for determination of the differences, and statistical significance was set at p < 0.05.
Results
Impaired antiviral immune response in NF-κB p52- but not in NF-κB p50-deficient mice
To investigate the ability of NF-κB p52- and NF-κB p50-deficient mice to control an LCMV infection, those mice and C57BL/6 wild-type animals were infected IV with 106 pfu LCMV-WE. Virus titer was determined at different time points after LCMV infection in blood (Fig. 1A) and spleens (Fig. 1B) of infected animals. Whereas LCMV was already detectable at 2 d p.i. in the blood of p50−/− mice (Fig. 1A, grey circles), the virus was first found in the blood of NF-κB p52−/− (Fig. 1A, open triangles) and wild-type (Fig. 1A, black squares) mice on day 4 p.i. At this time point LCMV reached maximum titers in wild-type (3.08 ± 0.33 log10 pfu/mL) and p50−/− mice (2.72 ± 0.02 log10 pfu/mL). Thereafter the amount of infectious virus in the blood of those animals slowly decreased. At 10 d p.i. no LCMV was detected in the blood of wild-type and p50-deficient mice. In sharp contrast, p52−/− mice were unable to eliminate the virus, and high viral loads (4.77 ± 0.65 log10 pfu/mL) persisted in the blood for as long as 130 d, when the experiment was terminated.
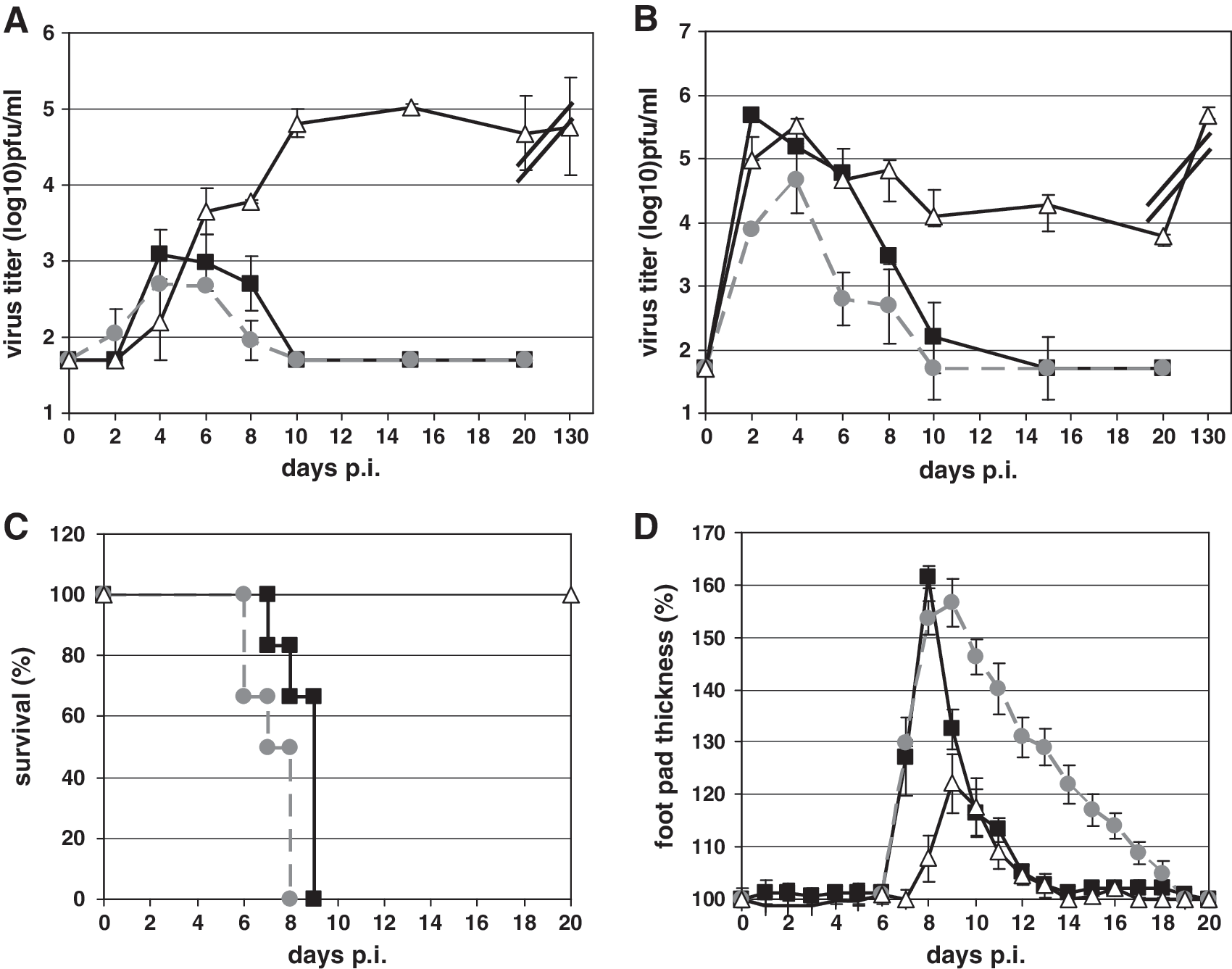
Determination of immune responses after different routes of LCMV infection with wild-type (black squares), NF-κB p50−/− (grey circles), and p52−/− (open triangles) mice. Kinetics of virus elimination are shown for blood (
A similar situation was observed in the spleens of LCMV-infected mice. As shown in Fig. 1B, at 2 d p.i. LCMV had replicated to high titers in wild-type (5.69 ± 0.06 log10 pfu/mL), NF-κB p50−/− (3.89 ± 0.52 log10 pfu/mL), and p52−/− mice (4.96 ± 0.37 log10 pfu/mL). Again wild-type (Fig. 1B, black squares) and NF-κB p50-deficient mice (Fig. 1B, grey circles) were able to eliminate the virus from the spleen within 15 d p.i. Again, no virus clearance was observed in p52-deficient mice (Fig. 1B, open triangles).
Intracerebral (IC) LCMV infection with 1 × 103 pfu of wild-type (Fig. 1C, black squares), as well as NF-κB p50−/− mice (Fig. 1C, grey circles), resulted in massive weight loss in association with the death of these animals between days 6 and 9 after LCMV inoculation of the brain. Surprisingly, NF-κB p52−/− mice survived the IC infection (Fig. 1C, open triangles). Those mice lost less than 20% of their body weight throughout the experiment, regained their full body mass after 30 d, and completely recovered from their clinical symptoms (data not shown).
LCMV inoculation into the footpad induces a local DTH reaction that results in footpad swelling that is sequentially mediated by class I-restricted cytotoxic CD8+ T cells and class II-restricted CD4+ T cells, cytokines and inflammatory cells (32,50). To investigate LCMV-mediated footpad swelling in wild-type, NF-κB p50−/−, and p52−/− mice, LCMV (1 × 104 pfu) was administered subcutaneously into the left hind foot. As seen in Fig. 1D, footpad swelling in wild-type mice was maximal between days 8 and 9 (Fig. 1D, black squares), and a subsequent decrease in footpad thickness was seen at day 9 p.i. Footpad swelling in p50−/− mice (Fig. 1D, grey circles) was similar to that of wild-type mice, although the maximum was reached at 10 days p.i. In NF-κB p52-deficient mice (Fig. 1D, open triangles) maximal footpad swelling was also seen at day 9 p.i., but the intensity was reduced threefold compared to wild-type animals. Taken together, all of the experiments using different routes of infection demonstrated no differences in the ability of NF-κB p50−/− mice to control LCMV infection compared to wild-type mice, whereas NF-κB p52-deficient mice were unable to control LCMV infection. To investigate the underlying mechanism behind the impaired immune response of NF-κB p52−/− mice in more detail, the following experiments were performed with wild-type and NF-κB p52−/− mice only.
Depletion of CD4+ T lymphocytes prevents DTH reaction in NF-κB p52-deficient mice
To analyze the impact of T-cell populations on the cellular immune response against LCMV in NF-κB p52-deficient mice in more detail, experiments were performed in which wild-type and NF-κB p52−/− mice were treated with antibodies to deplete either CD4+ or CD8+ T lymphocytes, or both immune cell populations. Immunocompetent untreated wild-type mice showed the classical swelling reaction, with peak swelling seen at 9 d p.i. (Fig. 2A, black squares), whereas the footpad swelling seen after LCMV infection of NF-κB p52−/− mice was fourfold lower (Fig. 2A, open triangles). Depletion of CD8+ T cells alone resulted in delayed and decreased swelling of the footpad in wild-type mice (Fig. 2B, black squares), as described earlier (50,51). In contrast, very little footpad swelling was observed in CD8+ T-cell depleted NF-κB p52−/− mice (Fig. 2B, open triangles). The depletion of CD4+ T cells in wild-type mice resulted in a diminished but not delayed swelling reaction (Fig. 2C, black squares). Maximal swelling was reached at 9 days p.i. Similarly to wild-type mice, CD4+ T-cell depletion in NF-κB p52−/− mice resulted in drastically attenuated footpad swelling (Fig. 2C, open triangles). Maximal swelling was seen at 11 d after virus inoculation, similarly to immunocompetent NF-κB p52−/− mice. Depletion of CD4+ plus CD8+ T cells in NF-κB-deficient mice resulted in complete suppression of footpad swelling (Fig. 2D, open squares), whereas a minimized swelling reaction was detectable in wild-type mice at 12 days p.i.
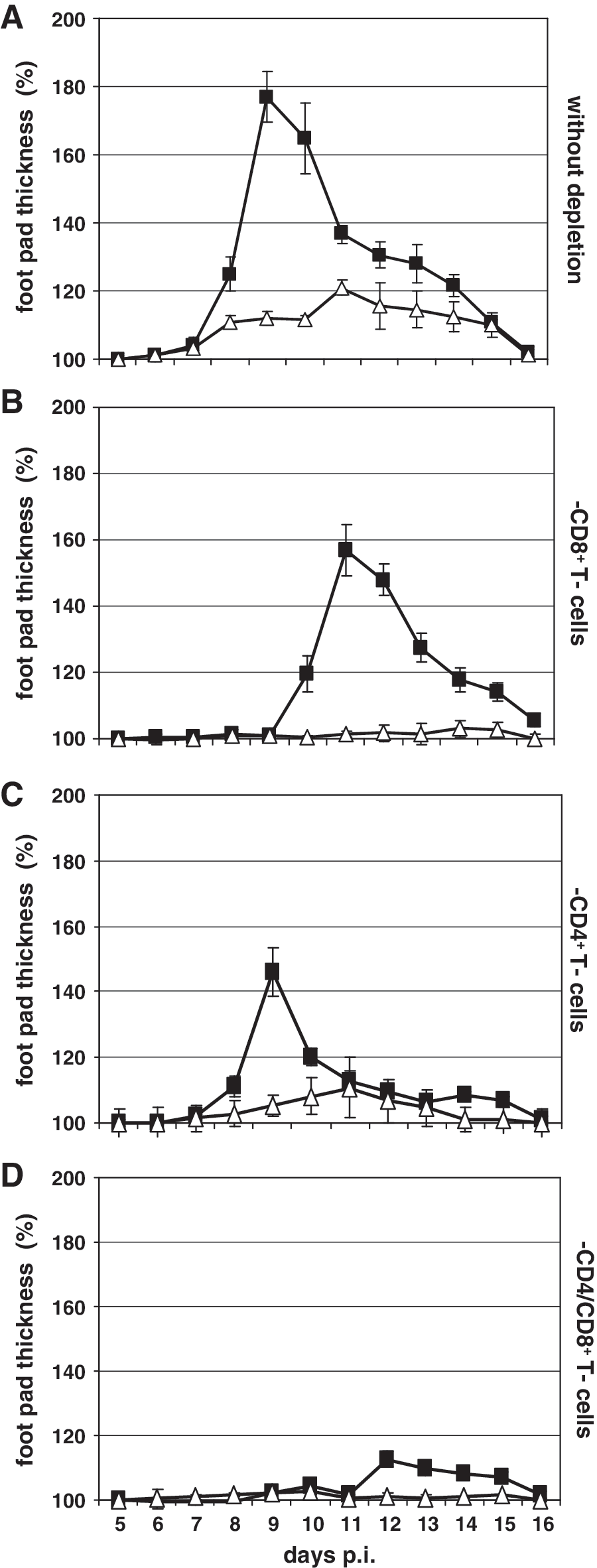
Footpad swelling reaction after depletion of T cells in wild-type and NF-κB p52−/− mice. (
Reduced cellular and humoral immune response in p52−/− mice after virus infection
The suppressed footpad swelling seen after LCMV infection in T-lymphocyte-depleted NF-κB p52−/− mice suggested that the function of both CD4+ and CD8+ T cells was impaired. To verify this finding, the cellular immune response mediated by cytotoxic antigen-specific CD8+ T cells in the spleens of wild-type and NF-κB p52−/− mice was investigated. By the use of a classical 51Cr-release assay as described in the materials and methods section, a 10-fold reduction in CTL activity was found in the spleens of NF-κB p52−/− mice (Fig. 3A, open triangles) 8 d after LCMV infection, compared to the CTL response seen in wild-type mice (Fig. 3A, black squares).
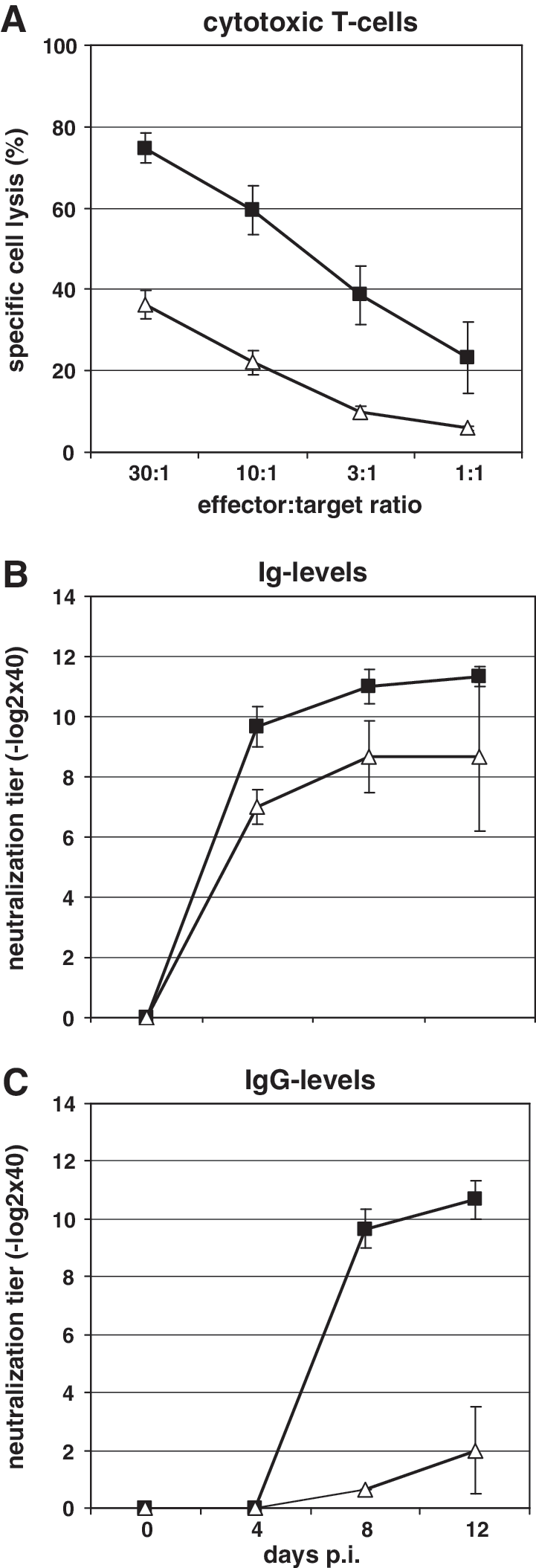
(
To investigate CD4+ T-cell activity in NF-κB p52−/− mice, we chose VSV infection as a model for CD4+ T-cell function analysis. VSV infection of immunocompetent mice induces a rapid T-cell-independent IgM response, followed by a long-lasting T-cell-dependent IgG response (34). Mice were infected with VSV-IND and bled at days 0, 4, 8, and 12 p.i. VSV-specific antibodies were found as early as 4 d p.i. (9.7 ± 0.7 −log2 × 40) in wild-type mice (Fig. 3B, black squares). The titer increased slightly at 8 (11.0 ± 0.6 −log2 × 40) and 12 d p.i. (11.3 ± 0.3 −log2 × 40). NF-κB p52−/− mice showed a similar titer of VSV-specific antibodies as that of wild-type mice (Fig. 3C, open triangles). Here VSV-specific immunoglobulins were also detectable at day 4 p.i. (7.0 ± 0.6 −log2 × 40), day 8 p.i. (8.7 ± 1.2 −log2 × 40), and day 12 p.i. (8.7 ± 2.5 −log2 × 40). In contrast, the overall amount of IgG-specific VSV antibodies was altered in NF-κB p52−/− compared to wild-type mice. Antibodies were first found at day 8 (9.7 ± 0.7 −log2 × 40), and the amount increased at day 12 p.i. (10.7 ± 0.7 −log2 × 40) in wild-type mice (Fig. 3B, black squares). In NF-κB p52-deficient mice, VSV-specific IgG antibodies were also found at days 8 and 12 p.i., but at low titers (0.6 ± 0.3 −log2 × 40 at day 8 p.i., and 2.6 ± 1.5 −log2 × 40 at day 20 p.i.). These data indicate that the humoral immune response against VSV in p52−/− mice is mediated by IgM, while the T-cell-dependent IgG response against VSV is impaired.
Since the LCMV-specific non-neutralizing humoral immune response is also T-cell dependent, we evaluated the humoral immune response against LCMV nucleoprotein (NP) by ELISA assay. Mice were infected with a high dose of LCMV (106 pfu) and were bled at days 0, 4, 8, and 20 p.i. The first IgM titers in wild-type mice appeared at day 4 p.i. (3.3 ± 0.3 −log2; Fig. 4A, black squares), and increased at day 8 p.i. to 5.7 ± 0.3 −log2. At day 20 p.i., the LCMV-specific IgM titer dropped to 4.3 ± 0.3 −log2. In NF-κB p52−/− mice the LCMV-specific IgM titer was almost 3 log2 higher at day 4 p.i. than the titer in wild-type mice (Fig. 4A, open triangles). The titer slightly decreased at day 8 p.i. (5.7 ± 0.3 −log2), and increased again at day 20 p.i. (6.5 ± 0.4 −log2). LCMV infection led to higher IgG titers in p52−/− mice at 4 days p.i. (7.7 ± 0.6 −log2; Fig. 4B, open triangles) compared wild-type mice (6.7 ± 0.3 −log2; Fig. 4B, black squares). The IgG titer in wild-type mice was increased at day 8 (10.0 ± 0.9 −log2) and day 20 p.i. (10.7 ± 0.7 −log2). In p52−/− animals a slight increase at day 8 p.i. was seen (8.0 ± 0.1 −log2), whereas the IgG titer was decreased at day 20 p.i. (6.0 ± 0.3 −log2; Fig. 4B, open triangles).
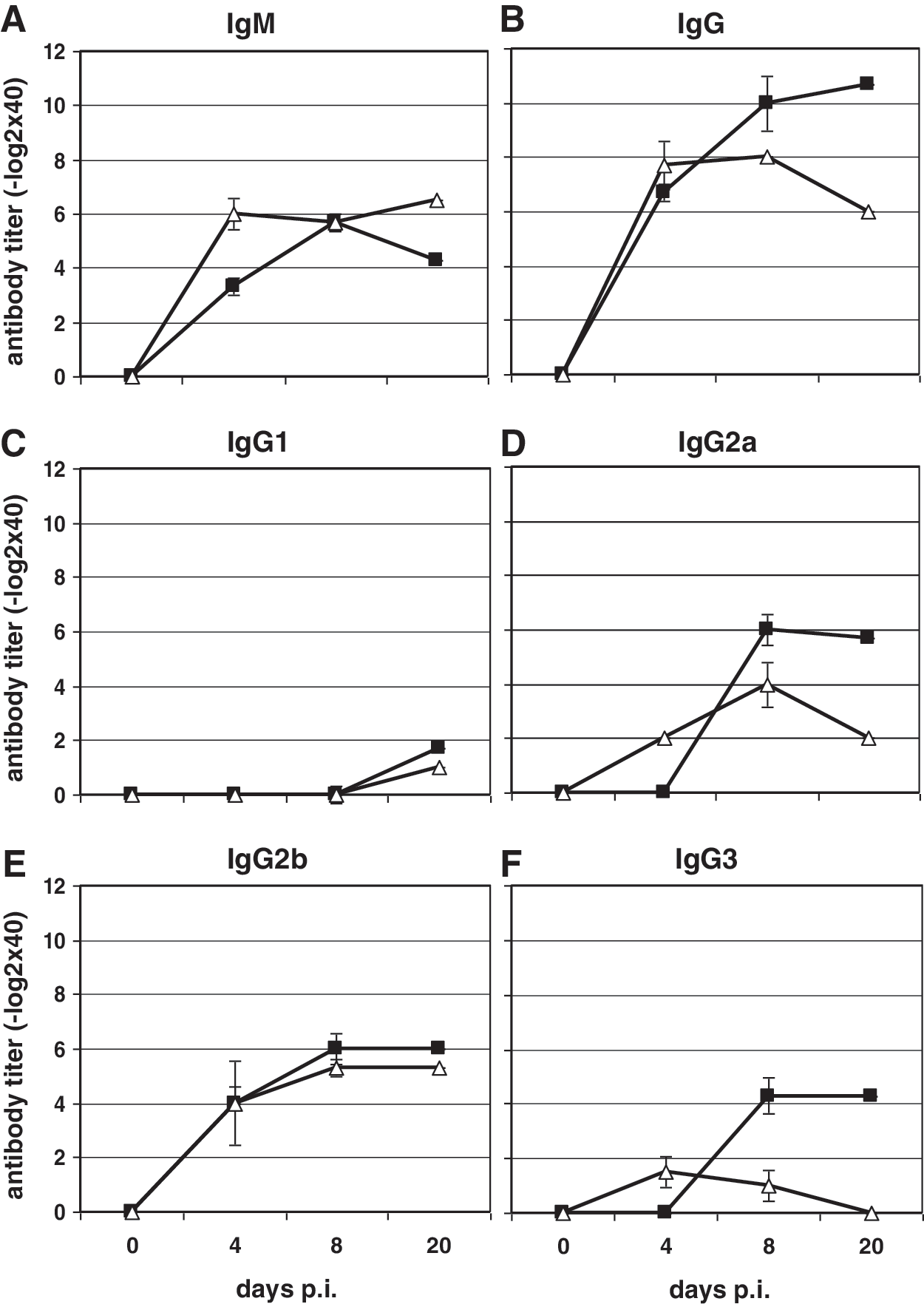
The humoral immune response seen 4, 8, and 20 d after LCMV infection (106 pfu) of wild-type and p52−/− mice. IgM (
The determination of IgG subclasses in the sera of LCMV-infected mice demonstrated almost no IgG1 response after LCMV infection in both mouse strains (Fig. 4C), while the titers of IgG2b (Fig. 4E) were almost equal in p52−/− and wild-type mice. In contrast, differences in the amounts of IgG2a and IgG3 were found between wild-type and NF-κB p52−/− mice (Fig. 4D and F). Even though p52−/− mice had higher IgG2a and IgG3 titers (2.0 ± 0.1 −log2 and 1.5 ± 0.6 −log2, respectively) at 4 d after LCMV infection compared to wild-type mice (no LCMV-specific antibodies were detectable), a reduction in these two IgG subclasses was observed in NF-κB p52-deficient mice at days 8 and 20 p.i. (IgG2a: 4.0 × 0.8 −log2 and 2.0 × 0.8 −log2; IgG3: 1.0 × 0.6−log2 and no antibodies) compared to wild-type animals (IgG2a: 6.0 × 0.6 −log2 and 5.7 × 0.3 −log2; IgG3: 4.3 × 0.7 −log2 and 4.3 × 0.7 −log2).
Reduced numbers of CD8+ T cells in the spleens of NF-κB p52-deficient mice after LCMV infection
To investigate the distribution of CD4+ and CD8+ T cells in wild-type and p52−/− mice after LCMV infection, immunohistochemistry of the spleens was performed. CD4+ T cells of wild-type mice were located within the T-cell zones of the splenic white pulp (Fig. 5A, white arrows). Four days after infection CD4+ T cells migrated out of the white pulp toward the red pulp (Fig. 5A, red arrows). Over the course of infection CD4+ T cells were detected within the white and red pulp in the spleens of wild-type mice. Twenty days p.i. distinct germinal centers within the white pulp of LCMV-infected wild-type mice were visible (Fig. 5A, black arrow). In the spleens of uninfected NF-κB p52−/− mice the CD4+ T cells were distributed within the white pulp, due to the lack of B- and T-cell-zone separation in the splenic microarchitecture (Fig. 5B, white arrows). The localization of CD4+ T cells in the spleens of NF-κB p52−/− mice did not change until day 8 p.i. Migration of CD4+ T cells out of the white pulp into the red pulp in the spleens of p52−/− mice was observed 8 d after LCMV infection (Fig. 5B, red arrow). Furthermore, no germinal center formation was detected in the spleens of LCMV-infected NF-κB p52−/− mice.

Immunohistochemistry of spleen sections of wild-type and NF-κB p52−/− mice after LCMV infection (106 pfu). (
The dissemination and amount of CD8+ T cells in the white pulp within the spleens of uninfected wild-type and NF-κB p52−/− mice revealed no differences (Fig. 5C and D, white arrows). At 4 d p.i. very few CD8+ T cells were present in the spleens of wild-type or NF-κB p52−/− mice (Fig. 5C and D, white arrows). Migration of CD8+ T cells out of the red pulp into the white pulp was observed at 8 d p.i. in wild-type mice (Fig. 5C, red and white arrows). In contrast, at the same time point very few CD8+ T cells were detected in the spleens of p52-deficient mice (Fig. 5D, red and white arrows). This same pattern was observed in the spleens of infected mice at 20 d p.i. Both wild-type and NF-κB p52−/− mice showed accumulations of CD8+ T cells within the white and red pulp at 20 d p.i.; however, the overall amount of CD8+ T cells in the spleens of NF-κB p52−/− animals was significantly reduced compared to the wild-type mice.
FACS analysis confirms reduced CD8+ T-cell numbers in NF-κB p52−/− mice
To further support the immunohistochemical observations, a quantitative analysis of CD4+ and CD8+ T lymphocytes in the spleens of LCMV-infected wild-type and p52−/− mice was performed by FACS analysis. In wild-type mice a 14.5% reduction in CD4+ T-cell numbers in the spleen was seen at 4 d after LCMV infection compared to uninfected animals (Fig. 6A, black bars). A similar reduction in CD4+ T-cell numbers (10%) was observed in the spleens of NF-κB p52−/− mice (Fig. 6A, white bars). At 8 d p.i. CD4+ T cell numbers were still reduced in wild-type mice, while a slight increase in these cells (15%) was found in NF-κB p52−/− mice (Fig. 6A, white bars). The amount of CD4+ T cells was increased at 20 d p.i. in wild-type mice, and nearly reached the level seen in uninfected controls. NF-κB p52−/− mice had similar numbers of CD4+ T cells at day 8 p.i. Significant differences in the amount of CD4+ T lymphocytes in the spleens of wild-type and NF-κB p52−/− mice were seen (p = 0.0069) at day 8 p.i.
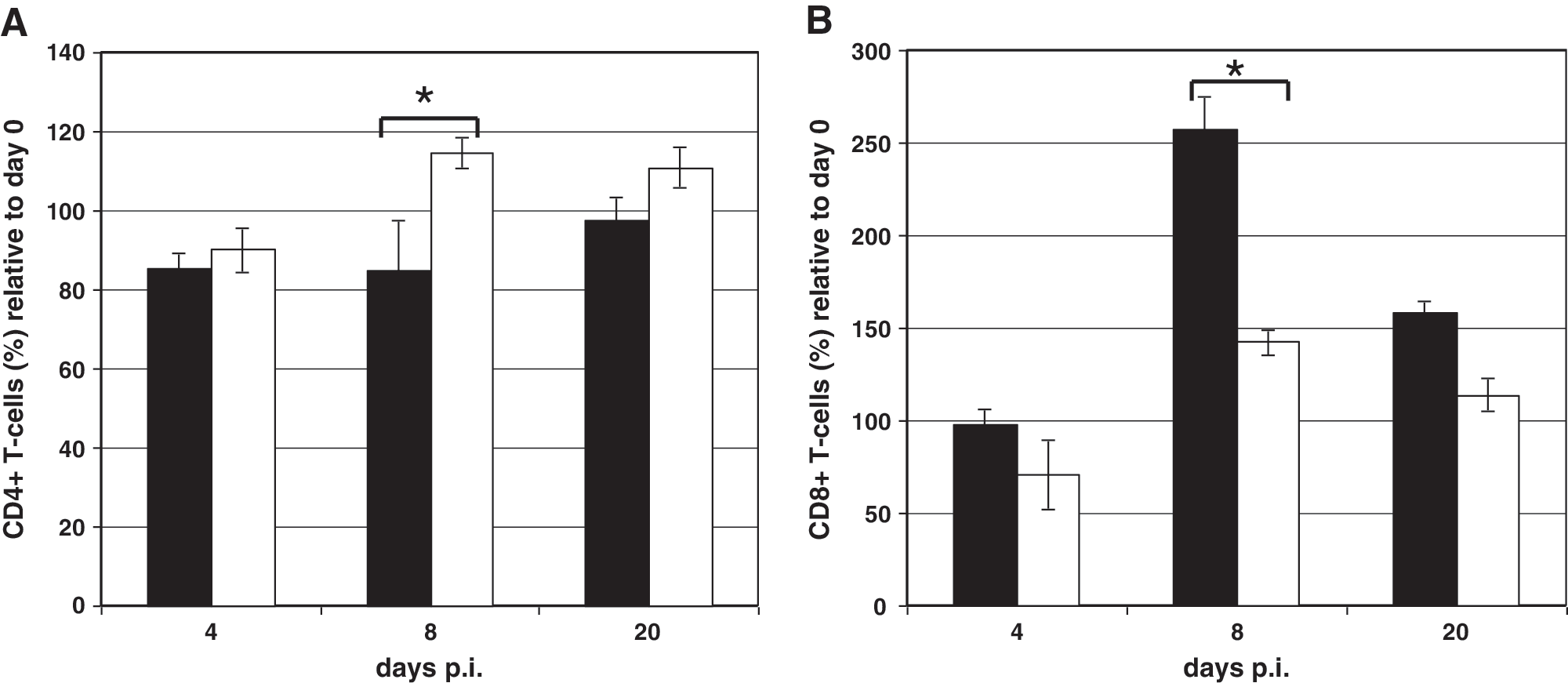
FACS analysis of spleen samples from wild-type and NF-κB p52−/− mice after LCMV infection (1 × 106 pfu). Wild-type (black bars) and p52−/− mice (white bars) were infected via IV injection into the tail vein. On days 0, 4, 8, and 20 p.i. the spleen cells were analyzed using the FACSCalibur and Cell Quest software. The amount of cells present at 4, 8, and 20 d p.i. is given as a relative percentage of the amount of cells present prior to infection. (
The numbers of CD8+ T cells present after LCMV infection in the spleens of NF-κB p52−/− mice was also altered compared to wild-type animals. At 4 d after LCMV infection in wild-type mice there was no difference in the amount of CD8+ T cells in the spleen compared to uninfected animals (Fig. 6B, black bars). NF-κB p52−/− mice showed a reduction (30%) of the CD8+ T-cell population at this time point (Fig. 6B, white bars). Clear differences (p = 0.0051) were observed at 8 d p.i. Wild-type mice showed a 160% increase in CD8-positive lymphocytes at this time point compared to uninfected controls (Fig. 6B, black bars). Although the amount of CD8+ T cells in NF-κB p52−/− mice was increased by 40% at 8 d p.i. compared to uninfected controls, the number of CD8+ T lymphocytes was drastically reduced (120%) compared to wild-type mice (Fig. 6B, white bars). The number of CD8+ T cells in wild-type mice (Fig. 6B, black bars) was decreased again at 20 d p.i., but was still elevated about 60% compared to uninfected animals. A decrease in the CD8+ T-cell population was also observed in the spleens of NF-κB p52−/− mice (15%) at 20 d p.i. These results confirmed the data obtained from immunohistochemistry, revealing differences in the amount and distribution of the CD8+ T-cell population in the spleens of NF-κB p52−/− mice after LCMV infection.
Lymphocytes from p52−/− mice are able to respond against a viral pathogen when transferred into wild-type recipients
It is well known that the splenic microarchitecture is impaired in mice lacking NF-κB family members. In NF-κB p50−/− mice, only the marginal zone B-cell population is impaired, while in p52−/− mice major defects in germinal centers, follicular dendritic cell (FDC) networks, and in the architecture of the marginal zone were found (7). We therefore questioned whether the observed inability of p52−/− mice to control LCMV infection is due to defects in the effector immune cell population, or due to defects in priming as a consequence of disrupted splenic microarchitecture.
Wild-type and NF-κB p52−/− recipient mice were thymectomized and treated with monoclonal antibodies to deplete the circulating CD4+ and CD8+ T cells as described in the materials and methods section. All recipient mice were assessed for successful depletion 2 wk later by FACS analysis. Thereafter, wild-type and NF-κB p52−/− recipient mice were reconstituted with naive lymphocytes from wild-type or NF-κB p52−/− sex-matched donors (Fig. 7). Two days later, all mice were challenged IC with LCMV (103 pfu). Wild-type mice, that either received lymphocytes from wild-type or p52−/− donor mice, developed disease and immunopathology after virus inoculation into the brain. In contrast, NF-κB p52−/− mice that either received lymphocytes from wild-type or p52−/− donor mice did not develop disease and became virus carriers (Table 1). These data indicate that mice lacking NF-κB family members of the alternative pathway show an impaired cellular immune response against viral infection, due to the disrupted splenic microarchitecture, but not due to impaired intracellular T-cell signalling.
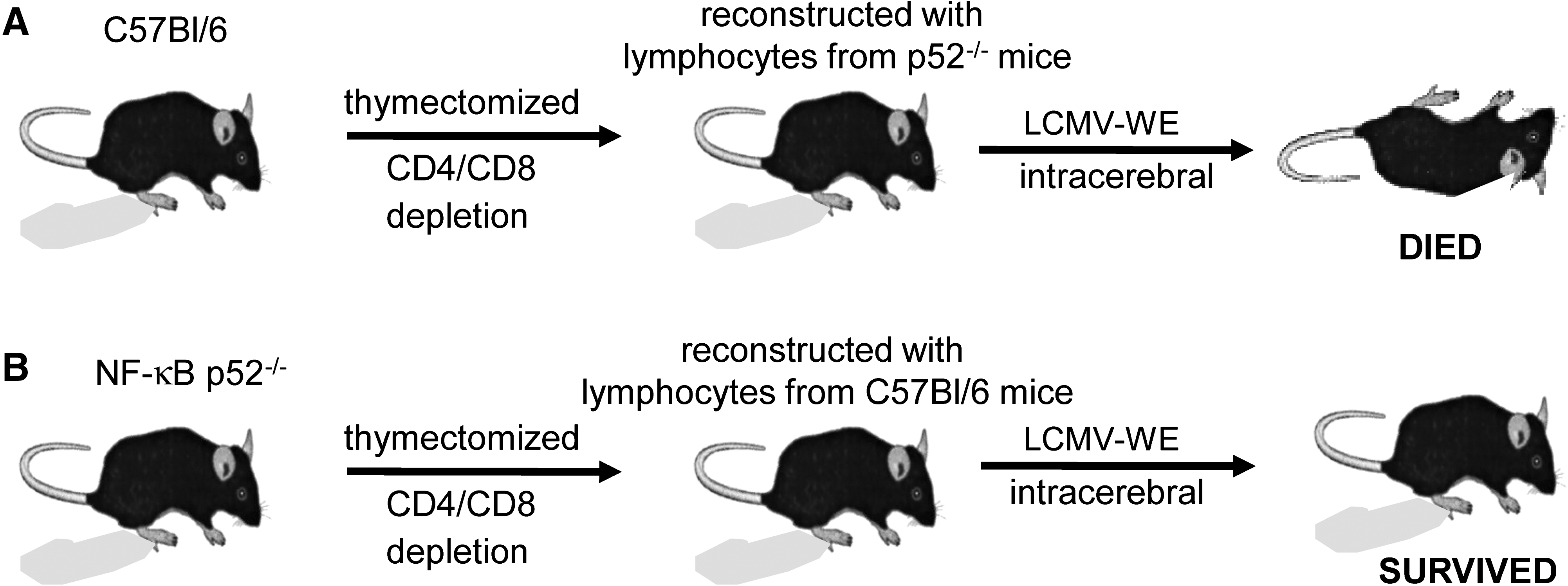
Adoptive transfer experiment in wild-type and NF-κB p52−/− mice. (
Virus titer is given as log10 pfu/mL organ homogenate.
T-cell chemoattractant CCL21 is nearly absent in p52−/− mice
It is well known that the alternative NF-κB signaling pathway is responsible for the regulation of the T-cell chemoattractant CCL21. Therefore we investigated whether the levels of CCL21 were altered in the spleens of wild-type and p52−/− mice compared to the CCL21 levels seen at 4, 8, and 20 d after LCMV infection. In uninfected wild-type mice 38 ± 11 ng CCL21/g spleen were detected (Fig. 8A, black circles). LCMV infection led to a decrease in CCL21 levels in the spleens of wild-type mice. At day 4 p.i. only 18 ± 3 ng CCL21/g spleen were found. Similar amounts were found at days 8 (18 ± 10 ng) and 20 p.i. (17 ± 3 ng). In the spleens of NF-κB p52−/− mice only small amounts of CCL21 were detectable. In uninfected p52−/− mice, 3 ± 0.6 ng/g of CCL21 were found (Fig. 8B, open circles). There were no changes in the CCL21 concentration after LCMV infection. At 4 d p.i. the spleens of p52−/− mice contained 3 ± 0.9 ng CCL21/g tissue, and the amount of the cytokine was unchanged at days 8 and 20 p.i. (4 ± 0.3 ng/g spleen). Taken together, these data indicate that downregulation of the T-cell chemoattractant CCL21 in p52−/− mice due to disrupted splenic microarchitecture might be responsible for the inadequate T-cell activity that was unable to control LCMV infection.
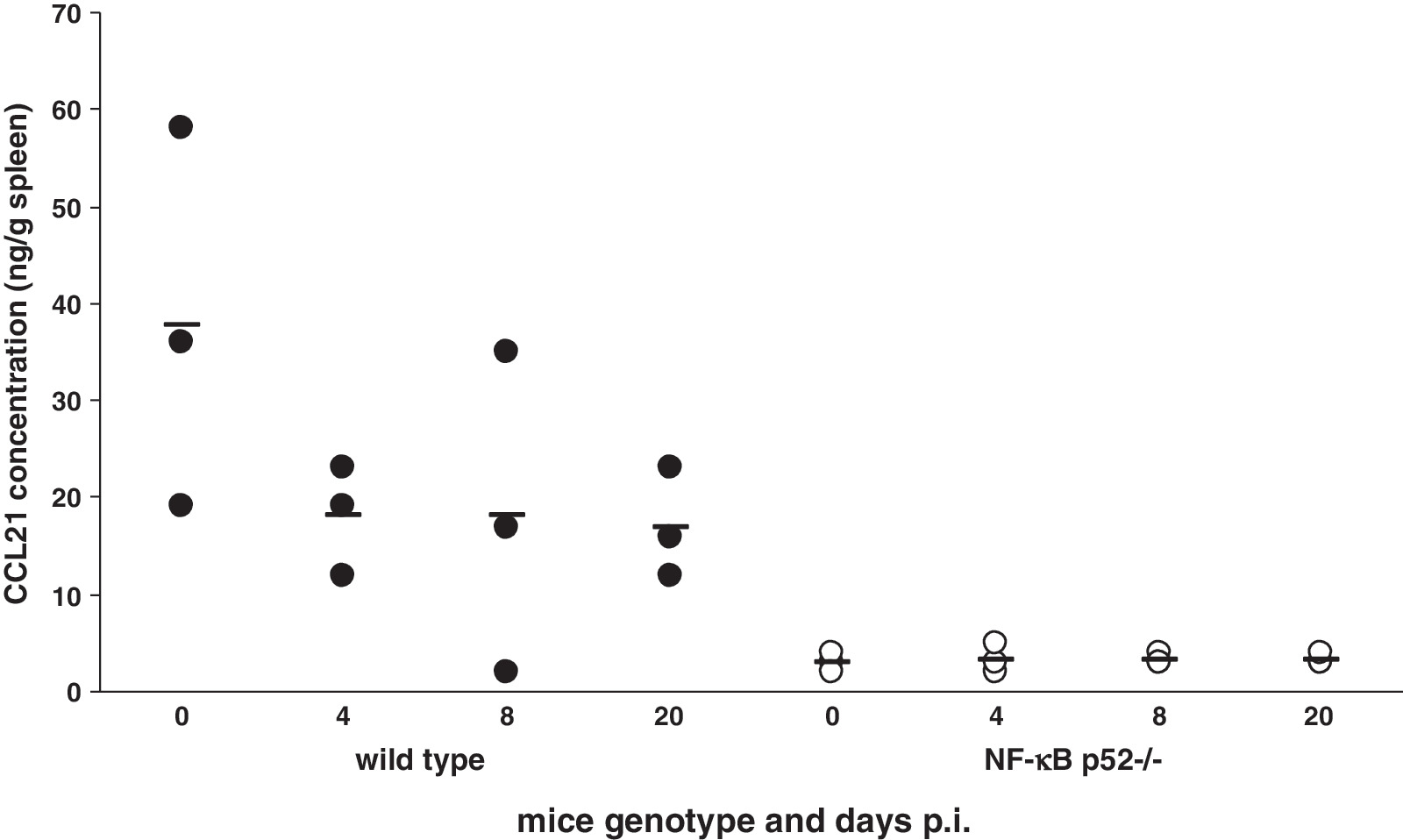
Concentration of CCL21 in the spleens of wild-type and NF-κB p52−/− mice. The amounts of the T-cell chemoattractant CCL21 in the spleens of uninfected and LCMV-infected wild-type (black circles) and NF-κB p52−/− mice (white circles) were investigated using ELISA. NF-κB p52−/− mice showed reduced amounts of CCL21 compared to wild-type animals. The results represent the median of two independent experiments with three mice per genotype.
Discussion
The data presented here demonstrate the importance of the alternative, but not the classical, NF-κB pathway in the defense against LCMV infection. NF-κB p52−/− mice were unable to control LCMV infection when infected by three different routes, whereas NF-κB p50−/− mice efficiently eliminated the virus. Peripheral LCMV infection usually stimulates a robust CD8+ T-cell response that is responsible for eliminating infected cells and controlling LCMV infection (21). This CTL immune response was 10-fold reduced in NF-κB p52−/− mice. These mice failed to clear LCMV and developed persistent infections. Supporting this finding, the CTL-mediated immunopathology in the brain seen after IC LCMV infection was absent in NF-κB p52-deficient mice. The intensity of the LCMV-mediated DTH reaction was also reduced. Depletion of participating T cells revealed not only reduced CD8+ T-cell activity, but also a defect in the CD4+ T-cell-mediated immune response, since the elimination of those cells prevented any measurable swelling reaction. The failure of the T-helper response was also confirmed by investigation of the humoral immune response after LCMV, and also after VSV infection. NF-κB p52−/− mice showed only slightly reduced Ig levels, but strongly impaired IgG responses, after VSV infection compared to wild-type mice. The analysis of the humoral immune response against LCMV nucleoprotein also demonstrated a reduced IgG response in NF-κB p52−/− mice, due to an attenuated IgG2a and IgG3 response. These results give rise to speculation that the class switch from IgM to IgG was affected in those animals. This might be due to the disrupted splenic microarchitecture seen in NF-κB p52−/− mice. The T-dependent B-cell response seen after antigen exposure infection is accompanied by the formation of germinal centers (37,66). Within these specialized microenvironments somatic mutations and affinity maturation take place (42). It was previously described that NF-κB p52−/− mice lack germinal centers (62). Furthermore, the disruption of the humoral immune response to T-cell-dependent antigens in those mice was previously demonstrated (17,62). In addition, NF-κB p52−/− mice were more susceptible than wild-type mice when infected with Toxoplasma gondii (11,13,17). The clearance of this parasite is a CD4+ T-cell-dependent process. The increased sensitivity to this parasite was explained by the insufficient activation of T-helper cells in NF-κB p52−/− mice. The data obtained in our study support these previous findings. However, the inability to control LCMV infection is primarily due to dysfunction of the cytotoxic T-cell response. One might speculate that the CTL response is impaired because of the lack of the CD4+ T-cell help that often sustains the CTL response (14,23,45). In fact, mice deficient in CD4 are unable to control LCMV infection solely with CTLs after high-dose injection, but these mice mounted a normal CTL response after low-dose inoculation (4). NF-κB p52−/− mice were unable to control a high-dose as well as a low-dose (data not shown) LCMV infection. The analysis of the cytotoxic activity of CTLs in the spleens of NF-κB p52−/− mice revealed a 10-fold reduction in activity compared to CTL activity in wild-type mice. Additionally, we found substantial differences in the numbers of CD8+ T cells present in the spleen after LCMV infection. In NF-κB p52−/− mice the number of CD8+ T cells was fourfold smaller than that of wild-type mice. From these data one might argue that a functional defect in the CTL response was responsible for these effects. However, the dysfunction of the cellular immune response after LCMV infection in NF-κB p52−/− mice was not directly due to a disorder of the CD8+ T lymphocytes, since lymphocytes from NF-κB p52−/− mice were effective when transferred into wild-type recipients. This indicates that the disturbed splenic microarchitecture of NF-κB p52−/− mice was responsible for the inadequate cellular immune response seen after LCMV infection. In this regard the finding that after LCMV infection CCL21 is nearly completely absent in NF-κB p52−/− mice supports the above hypothesis. CCL21 is a T-cell chemoattractant that is associated with modulation of immune responses. CCL21 is secreted by the T-cell stromal cells and it functions as a ligand for the CCR7 that is expressed on T cells. CCL19 and CCL21 are crucial for the positioning of T cells and dendritic cells (DC) within the T-cell zones of secondary lymphoid organs (43,44, 67). Interestingly and in contrast to results of an earlier study using RAG-deficient mice (52), the virus titer in the disrupted spleens of NF-κB-deficient mice was not reduced (Fig. 1B) compared to wild-type mice.
The splenic marginal zone is composed of metallophilic and marginal zone macrophages (28). Splenic marginal zone macrophages have been previously shown to be an essential cellular component for the clearance of LCMV (65). However, the spleens of NF-κB p52−/− mice contain marginal zone macrophages (62), and these cells are not required for the induction of cytotoxic T-cell responses (1). The highly-organized microarchitecture of the spleen and the interactions between components of the innate and adaptive immune systems can generate an efficient immune response. The priming of naive B and T cells occurs within the spleen (25). DCs play a major role in the activation of innate and adaptive immunity (2,29,30). These specialized antigen-presenting cells are able to induce antiviral cytotoxic T-lymphocyte responses (26,38,41). One of the factors determining the effectiveness of the T-cell response is the maturation status of the DCs. This maturation is highly dependent on the transcription factor NF-κB (68,69,71). Moreover, two recent articles highlighted the essential involvement of the alternative NF-κB pathway in maturation and T-cell priming of DCs (36,49).
Conclusion
We were able to demonstrate that the NF-κB p52 subunit is crucial for an adequate CTL response against LCMV. The underlying mechanism involves defects in maturation and priming rather than impaired intracellular signalling in CD8+ T cells. The absence of NF-κB p52 leads not only to severe defects in lymphoid organs, but it may also be essential for the defense against viral pathogens. The understanding of the effects caused by the modulation of multiple adaptive immune responses through NF-κB impairment may aid in the development of new strategies for the prevention of autoimmune diseases. Manipulation of the immune system via the NF-κB pathway may also be beneficial for tumor vaccine strategies. Moreover, it has been shown that targeting cellular factors like the classical NF-κB pathway as an antiviral strategy is a promising new treatment approach (39,46,63). The results reported here further support this strategy, since mice with defects in the classical NF-κB signalling pathway (p50−/− mice) were still able to mount an effective cellular immune response against LCMV.
Footnotes
Acknowledgments
We would like to thank Katja Oesterle, Katja Flint, and Ulrich Wulle for their excellent technical assistance. Furthermore, we would like to thank R.M. Zinkernagel for critical discussion and analysis. This work is part of the EUROFLU consortium activities, and of the VIRGIL European Network of Excellence on Antiviral Drug Resistance, supported by a grant (LSHMCT-2004-503359) from the Priority 1 “Life Sciences, Genomics and Biotechnology for Health” program in the 6th Framework Program of the European Union. This research was also partially supported by the Federal Government of Germany under the Influenza Research Programme “FSI,” and by the Bundesministerium Für Bildung Und Forschung (BMBF) Zoonose program “FluResearchNet.”
Author Disclosure Statement
No competing financial interests exist.