
Abstract
Impaired antiviral CD8 and CD4 T-cell responses are often associated with chronic viral infections. Cell-intrinsic as well as cell-extrinsic mechanisms are thought to dampen such responses, for example programmed death 1 receptor (PD-1) expression on T cells, and interleukin (IL)-10 production primarily by dendritic cells (DCs), have been shown to support viral persistence by suppressing immune responses. Here we demonstrate that CD103, an alpha E integrin necessary for T-cell homing and retention in the gut and other epithelia expressed by the majority of naïve CD8+, and CD4+CD25+ T cells and some DC subsets, is unnecessary for controlling T-cell responses during chronic lymphocytic choriomeningitis virus clone 13 (LCMV cl13) infection. T-cell analysis following viral infection showed that the primary as well as the memory CD8+ and CD4+ T-cell responses among CD103-sufficient and CD103-deficient mice were identical. In addition, no rescue of cytokine production by virus-specific T cells or alterations in viral titers in the absence of intrinsic CD103 expression was observed. Interestingly, CD103 levels on the effector CD8+ T cells became reduced soon after virus infection, with a small proportion of cells co-expressing PD-1 and CD103. In contrast, although no substantial differences in the frequency and number of the CD4+CD25+ cell population were seen, CD103 expression increased significantly over time in this population, correlating with viral persistence. Thus, a lack of CD103 expression does not affect functional impairment of effector T-cell responses during chronic viral infection.
Introduction
Regulation of the host antiviral immune response can occur at different levels. CD4+CD25+ T regulatory cells (Tregs) have been shown to control virus-specific primary and memory CD8+ T-cell responses (10 –12). Such control occurs either by directly inhibiting their functions, or indirectly modulating the activity of antigen-presenting cells (APCs) (13,14). Recently, in a mouse model of chronic infection with Leishmania major, E-cadherin, the primary ligand of CD103 (αEβ7) (15,16), which is highly expressed at the surface of keratinocytes and Langerhans cells in the skin, was found to contribute to disease outcome. In this study, genetically susceptible BALB/c mice that lacked CD103 became resistant to infection, a phenotype that was associated with a poor capacity of CD103-expressing Tregs to be retained at the infected site (17). Although CD103 is not a homing receptor, the ability of CD103 to bind to E-cadherin–expressing epithelial cells is thought to retain lymphocytes at the epithelial surface, playing important roles in T-cell homing to the skin and other tissues, primarily within the gut (18,19). The contribution of CD103 to gut-associated trafficking was also shown in studies involving pancreatic islet allo-transplantation, in which allografts survived indefinitely in CD103-deficient hosts (20).
To gain further insight into the role of CD103 in the establishment and maintenance of prolonged viral infections, we investigated whether this integrin is involved in the persistence in a murine viral infection model. LCMV is an arenavirus that can cause either acute (Armstrong-clone 53b) or persistent/chronic infection (clone 13) in vivo, depending the mouse strain, route of infection, and dose of the virus (21). Adult mice infected with LCMV Armstrong rapidly clear the infection and establish a stable memory T-cell pool (22 –24), while infection with the naturally selected isolate clone 13 results in chronic disease (25 –27). Immunologically one finds lymphopenia, functional T-cell impairment or “exhaustion,” and deletion of some virus-specific CD8+ and CD4+ T cells (28 –30), resulting in viral persistence, which was recently linked to expression of the programmed death 1 receptor (PD-1), and the expression of immunoregulatory cytokines such as IL-10 and IL-21 (31 –35).
Previously, we showed that CD103 plays a minimal role in a virally-induced model of type 1 diabetes (36). In this model, CD103-deficient mice crossed with rat insulin promoter-lymphocytic choriomeningitis virus (RIP-LCMV)-transgenic mice were monitored for type 1 diabetes development after acute infection with LCMV Armstrong. It was found that CD103 deficiency had no significant effect on disease progression, or on the anti-LCMV-specific effector and memory CD8+ or CD4+ responses. These results provided evidence for the clinical safety of CD103 blockade in potential future immune intervention trials. However, the role of CD103 during chronic infections, as they can be expected to present in humans, was never addressed. In the present study, although CD103 expression on CD4+CD25+ T cells strongly correlated with viral persistence, its ablation did not significantly alter the antiviral immune response. Therefore, targeting CD103 to treat persistent viral infections would be of no therapeutic value; however, it should be safe for interventions in other immune-based disorders, such as after islet allo-transplantation or Leishmania major infection.
Materials and Methods
Mice
Mice deficient in CD103 (CD103−/−) were kindly provided by Schon et al. (37). CD103−/− mice were back-crossed on a C57BL/6 background and CD103+/− mice were intercrossed to obtain CD103+/+, CD103+/−, and CD103−/− animals. In some experiments, RIP-GP × CD103−/−, or CD103+/− and CD103+/+ mice were used (36). The mice were housed under specific pathogen-free conditions at the La Jolla Institute for Allergy and Immunology.
Viruses
LCMV Armstrong (clone 53b) or clone 13 were used throughout all experiments. Virus stocks were prepared as previously described (31,38). Mice 8 to 14 weeks old were infected with a single dose of 104 plaque-forming units (PFU) intraperitoneally, or 2 × 106 PFU intravenously of each virus, respectively.
Peptides
The peptides used for the viral studies were the dominant Db-restricted LCMV epitopes GP33–41 (GP33) and NP396–404 (NP396) as well as an I-Ab–restricted epitope GP61–80 (GP61) (all from Abgent, San Diego, CA).
Flow cytometry
The following fluorescein isothiocyanate (FITC)-, phycoerythrin (PE)-, PE-Cy7-, peridinin chlorophyll-a protein (PerCP5.5)-, allophycocyanin (APC)-, APC-Cy7, and pacific blue (PB)-conjugated antibodies (BD Biosciences Pharmingen, Franklin Lakes, NJ or eBioscience, San Diego, CA) were used for flow cytometry for CD4, CD8α, CD44, CD25, CD62L, PD-1, and CD103, respectively. DbGP33 and DbNP396 class I pentamers were obtained as PE conjugates from Proimmune Inc. (Bradenton, FL) and used as described previously (31,36,38).
For intracellular stains, single-cell suspensions were restimulated for 3 h with 1 μg/mL MHC class I-restricted viral peptides, 2 μg/mL MHC class II-restricted viral peptides in the presence of brefeldin A (Sigma-Aldrich, St. Louis, MO). The cells were stained for surface expression of CD4 and CD8, fixed, permeabilized with the BD-Pharmingen Cytokine-Cytofix kit, and stained for intracellular IFN-γ, IL-2, and TNF. Cells were acquired on a LSRII flow cytometer (BD Biosciences Pharmingen) and analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
LCMV plaque assay
Organs (kidney and liver) were snap-frozen, weighed, and homogenized. Next, five different dilutions of each homogenized organ were prepared and plated on six-well plates coated with Vero cells as previously described (31). After the wells were stained with neutral red, they were treated with 3.7% formaldehyde. The infectious centers were counted, and the counts were averaged per group PFU. Viral LCMV stock was used as a positive control.
Statistical analysis
Data are expressed as mean ± SD. The statistical significance of the difference between means was determined using the two-tailed Student's t-test (*p < 0.05, **p < 0.001, and ***p < 0.005).
Results
Persistent infection with LCMV cl13 increases CD103 expression on CD4+CD25+ T cells
CD103 expression on Tregs was shown to contribute to their retention at the site of Leishmania infection and therefore susceptibility to the disease (17). This observation prompted us to investigate the role of CD103 (the αE chain of the αEβ7 integrin) in chronic LCMV cl13 infection. Initially, we followed the expression of CD103 in the blood, spleen, and mesenteric lymph nodes (mLN) during the course of LCMV Armstrong (clone 53b) or cl13 infection in C57BL/6 mice. We found that CD103 was differentially expressed on CD4+CD25+ T cells in all three lymphoid compartments after cl13 infection (Fig. 1A and B; data not shown for blood). In naïve mice, a significant proportion of CD4+CD25+ T cells expressed CD103: on average 20% in the spleen and blood and 40% in the mLN. Notably, in C57BL/6 mice infected with LCMV cl13 and not Armstrong, CD103 levels within the CD4+CD25+ T-cell population increased after the acute phase (day 8 post-infection [p.i.]), and reached 50% in the spleen and >60% in the mLN at the chronic phase of infection (day >45 p.i.). This change in CD103 expression levels suggests that either upregulation of the CD103 expression took place, and/or indicates changes in CD4+CD25+ T cells' homing ability. Additional analysis that included co-staining for the Treg transcription factor Foxp3 showed that on average 60% of the CD4+CD25+ T cells are Tregs, and that a majority express CD103 (Supplementary Fig. 1; see online supplementary material at
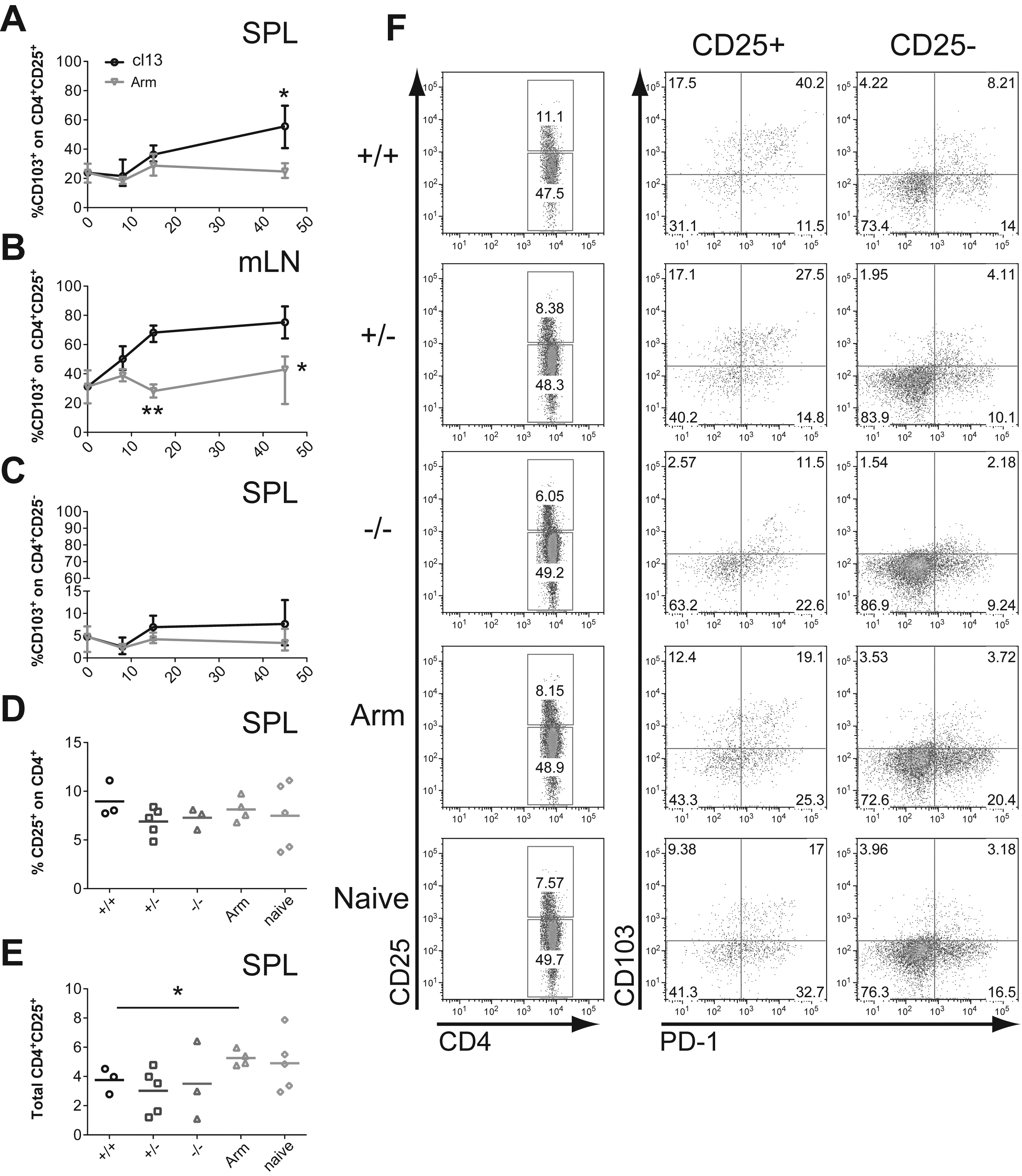
The expression of CD103 is highly upregulated after LCMV cl13 infection on CD4+CD25+ Tregs. C57BL/6 CD103+/+ mice were infected intravenously with 2 × 106 PFU LCMV cl13 or 104 PFU LCMV Armstrong (Arm) intraperitoneally. At different time points following infection the expression of CD103 at the surface of CD4+CD25+ cells in the spleen (
When the frequency and total CD4+CD25+ T-cell numbers between CD103 wild-type, CD4+CD25−, and CD4−CD25− LCMV cl13-infected mice were compared in the spleen 45 d after infection, no significant differences were found (Fig. 1D and E). By contrast, CD4+CD25+ splenocyte numbers were found to be reduced compared to LCMV Armstrong, due to cl13-induced lymphopenia (Fig. 1E). Notably, a majority of CD4+CD25+ T cells expressing CD103 co-expressed PD-1 in chronically infected mice at day >45 p.i. in the spleen and mLN (Fig. 1F). In summary, the expression of CD103 on CD4+CD25+ Tregs appeared to gradually increase over the course of the chronic cl13 infection.
CD103 expression is downregulated on effector CD8+ T cells over the course of a chronic viral infection
We examined the surface expression of CD103 on CD8+ T cells isolated from the spleen, blood, and mLN from mice infected with LCMV Armstrong or cl13 (Fig. 2A and B, and data not shown) at the peak (day 8), contraction (day 15), and late/persistent (day >45) phase of the anti-LCMV response in chronically infected mice. Interestingly, and in contrast to the Treg population, on average 60% of CD8+ lymphocytes expressed CD103 in all lymphoid compartments in naïve mice, while at the peak and contraction phases of the antiviral response, independent of the virus strain used, a massive reduction was seen. At these two time points, few CD103-expressing CD8+ T cells were detectable only in the mLN of LCMV Armstrong (acutely)-infected mice (Fig. 2B). At later time points an increase in CD103 expression levels was observed, which again did not diverge among the different LCMV strains.
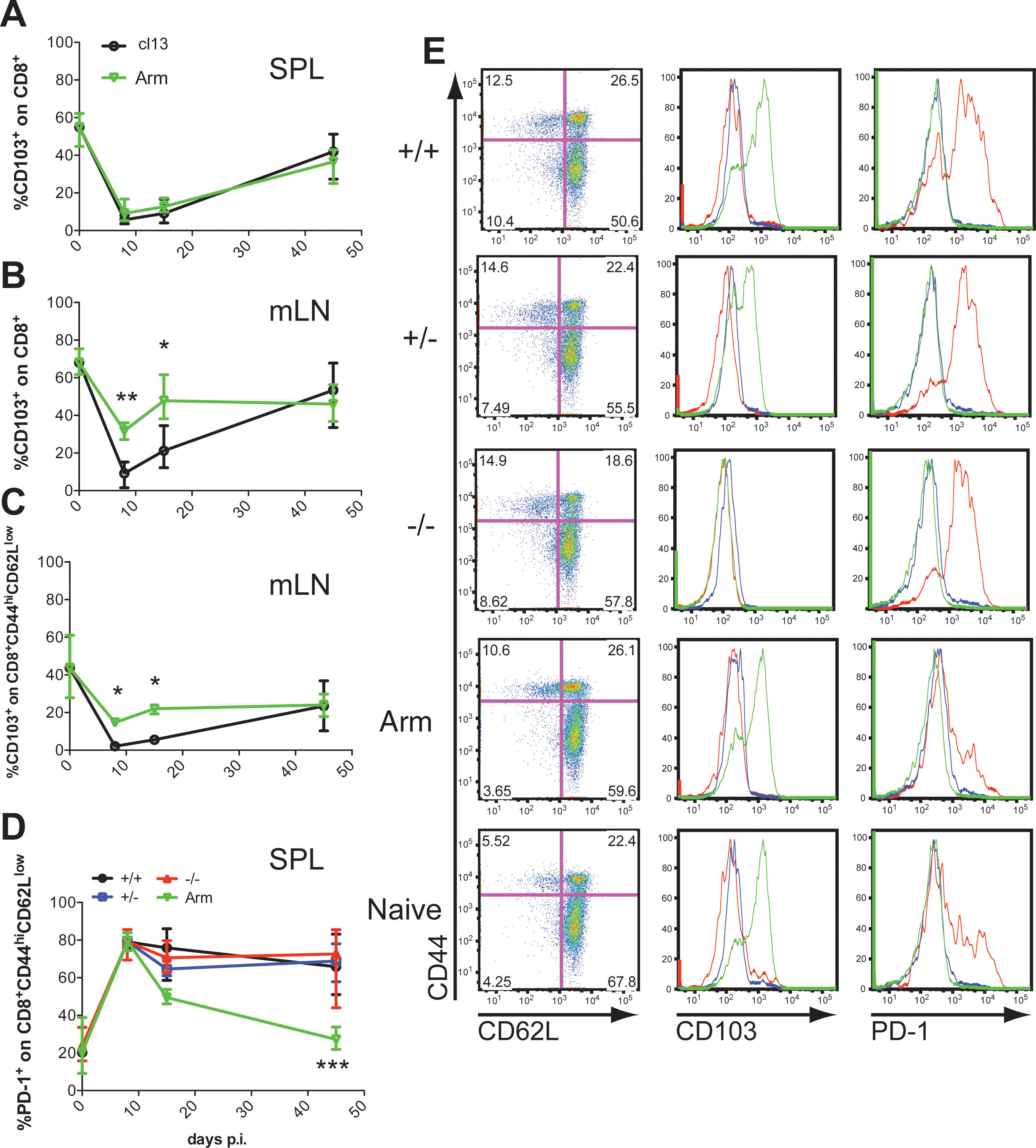
Significant reduction in CD103 levels on CD8+ T cells at early stages during the anti-LCMV immune response. C57BL/6 CD103+/+ mice were infected intravenously with 2 × 106 PFU LCMV cl13 or 104 PFU LCMV (Arm, Armstrong) intraperitoneally. At 8, 15, and >45 days p.i., the frequency of CD8+ lymphocytes that expressed CD103 in the spleen (
Next, CD103 levels within the effector memory (CD62LlowCD44high) CD8+ T-cell population were determined. The majority of CD8+CD62LlowCD44high cells did not express CD103 in the spleen, but significantly higher levels were seen in the mLN at a naïve state (Fig. 2C and data not shown). Upon LCMV infection, the CD103 expression within this population decreased further and increased only at later time points, following similar kinetics as those described for the entire CD8+ population. Again, the decline in CD103 levels was more pronounced after LCMV cl13 than with Armstrong infection (Fig. 2C). To better define the potential for CD103 during chronic infection, PD-1 levels within the effector memory CD8+ T-cell population were followed at different time points in CD103+/+, CD103+/−, and CD103−/− mice. As previously described, PD-1 levels did not decline upon LCMV cl13 infection in CD103+/+ mice, which is characteristic for exhausted CD8+ T cells (39 –41). However, as shown in Fig. 2D, no differences were seen between any of the CD103 groups. In order to better assess the population of cells expressing CD103 within the CD8+ T cells, a histogram overlay was conducted for spleen cells derived from LCMV-infected or naïve mice, after gating on the naïve, effector memory, and central memory CD8+ populations. Interestingly, as shown in Fig. 2E, a majority of cells that express CD103 fell within the naïve CD8+ T-cell population, suggesting a potential role for this molecule in the retention of naïve cells in different lymphoid compartments.
CD103−/− mice develop chronic infection with LCMV cl13
The sustained expression of CD103 on CD4+CD25+ Tregs in LCMV cl13-infected mice suggested that impairment of the expression of this molecule might affect their function. To determine the impact of CD103 ablation, we infected wild-type, CD103+/−, and deficient CD103 C57BL/6 mice intravenously with 2 × 106 PFU LCMV cl13. This dose and route induces chronic infection, which is associated with reductions in body weight, spleen cell numbers, and sustained high viral titers. Monitoring weight loss among the groups revealed no difference between wild-type and CD103-deficient mice (Fig. 3), and both wild-type and CD103−/− mice displayed significant and almost identical weight loss in both sexes (Fig. 3A and B). Among the females one mouse in each group did not survive the infection. Interestingly, wild-type males showed a much greater weight loss at day 15 p.i. than the CD103+/− and CD103−/− males.

CD103−/− C57BL/6 mice develop chronic infection after inoculation with the LCMV cl13 variant. Wild-type, CD103+/−, and CD103−/− C57BL/6 mice were infected intravenously with 2 × 106 PFU LCMV cl13, whereas control wild-type mice were infected with 104 PFU LCMV Armstrong (Arm) intraperitoneally. Weight loss was monitored over time. Values represent the mean of weight loss in female (
Viral load in the kidney and liver was also determined using plaque assays at 8, 15, and >45 days p.i. As shown in Fig. 4A and B, significant but similar reductions in viral load in both tissues were seen after day 8 p.i. and beyond. As expected, virus could still be detected in mice from all groups even at day 45 p.i. No significant differences in the kinetics of viral clearance between groups were detected. As shown in Fig. 4C and D, by >45 days p.i., 50% of the wild-type, 40% of the CD103+/−, and 67% of the CD103−/− mice had cleared the virus from the liver, while very few mice in all groups had also cleared the virus from the kidney. In summary, these results demonstrate that CD103 is not important for the regulation of the immune response during systemic chronic viral infection.
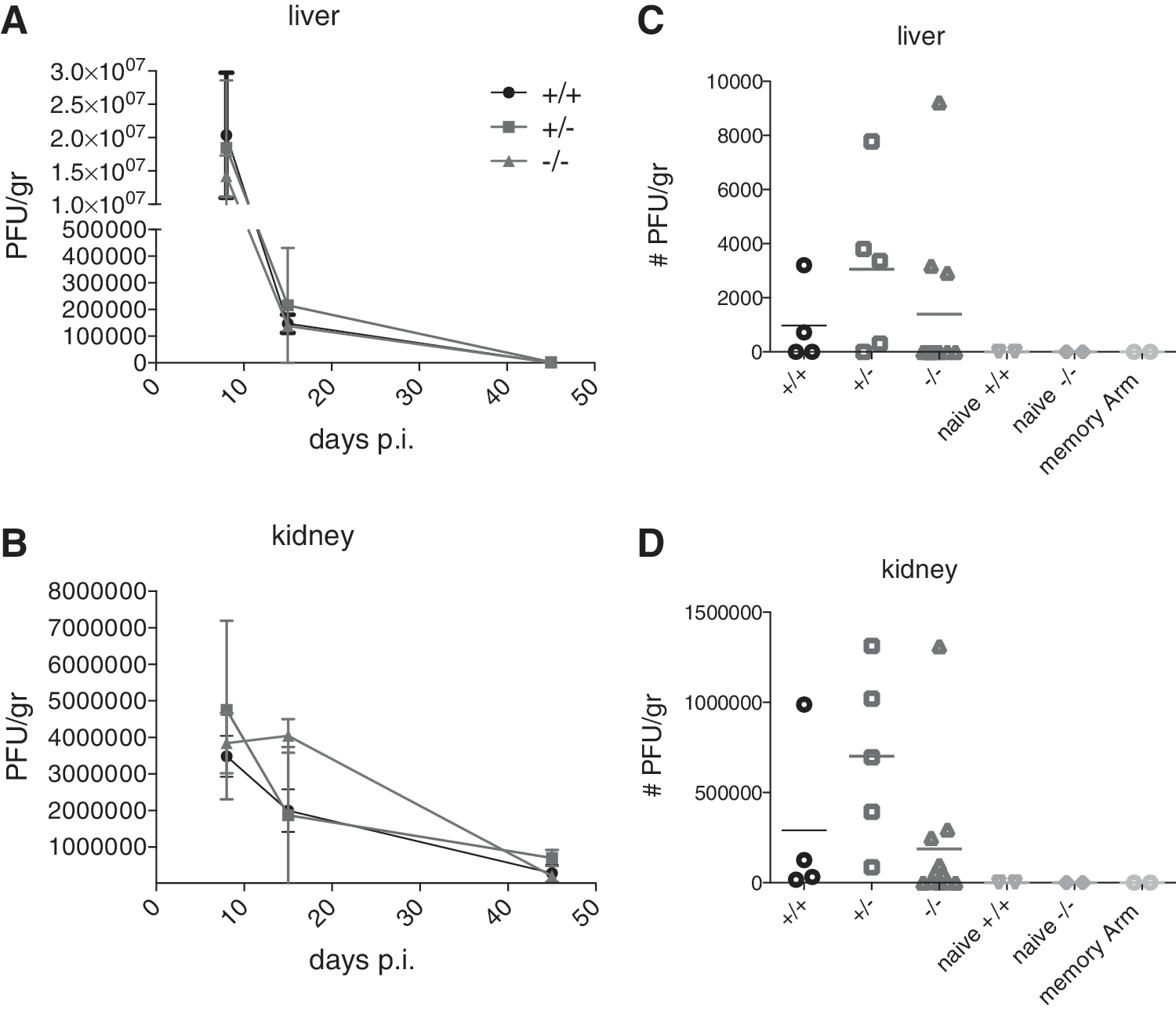
Similar LCMV cl13 virus titers are found in LCMV Cl13-infected CD103−/− and wild-type mice. Mice were infected with 2 × 106 PFU LCMV cl13 intravenously, and at 8, 15, and >45 days p.i., the number of PFU per gram in liver (
Primary and memory anti-LCMV cl13 responses are similarly impaired in CD103 +/− and wild-type mice
As reported previously (35,42), chronic infection with LCMV cl13 is associated with functional impairment and deletion of virus-specific CD8+ and CD4+ T cells, leading to a drastic decrease in the number of spleen cells (lymphopenia). In this study, analysis of cell counts from the spleen and mLN of the infected mice, regardless of the endogenous CD103 expression, showed similar signs of lymphopenia (data not shown). Next, we assessed the effect of CD103 on the LCMV-specific T-cell response. Splenocytes from CD103 wild-type, CD103+/−, and deficient mice were isolated at various time points after LCMV cl13 infection and stained with the MHC class I-specific pentamers GP33 and NP396. For control, C57BL/6 mice were infected with LCMV Armstrong. As shown in Fig. 5A and B, similar numbers of CD8-GP33 and NP396 pentamer-positive T cells were found in the spleen of LCMV cl13-infected mice regardless of endogenous CD103 expression, and at much lower numbers than in LCMV Armstrong-infected mice.
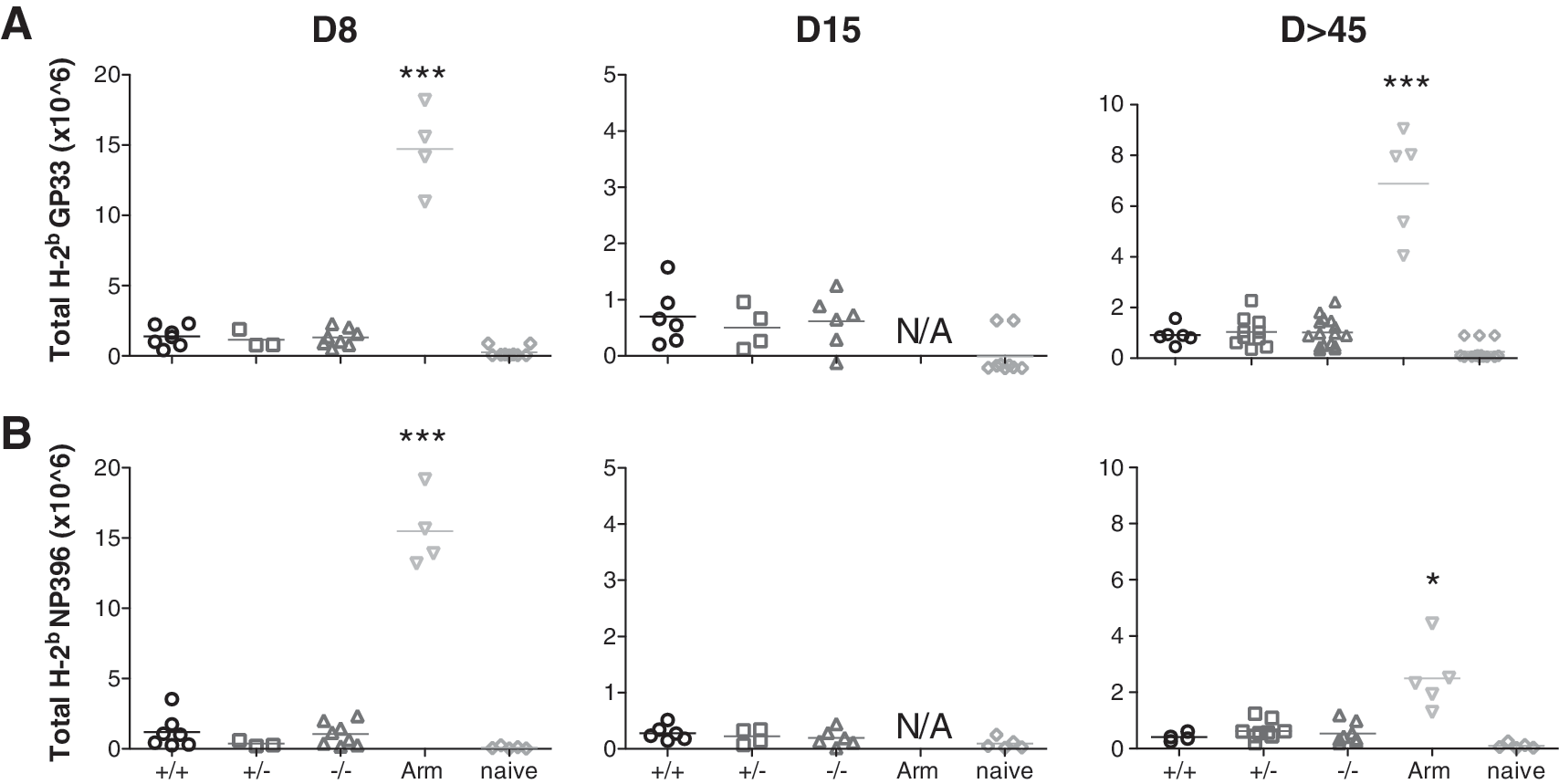
No effect was seen on overall impaired LCMV-specific CD8+ T-cell numbers through the absence of CD103 in persistent infection. CD103+/+, CD103+/−, and CD103−/− mice were infected with 2 × 106 PFU LCMV cl13 intravenously, and spleens were collected and analyzed at 8, 15, and >45 days after infection. For control, wild-type mice were infected with 104 PFU LCMV Armstrong intraperitoneally. Cells were stained for the presence of GP33 and NP396 pentamers and analyzed by flow cytometry. The total number of GP33 and NP396 cell numbers were calculated after multiplication of their frequency with their respective total spleen cell number. The GP33 (
In addition, intracellular cytokine staining following stimulation with class I- and class II-restricted immunodominant LCMV peptides GP33/NP396 and GP61 (data not shown) showed no differences in IFN-γ/IL-2 production at early time points (day 8 and day 15 p.i.), and IFN-γ production at later time points (day >45) (Fig. 6). Similarly to what has been described as an exhausted CD8+ T-cell phenotype, no significant IL-2 production could be detected in either group at day >45 p.i. (data not shown) except the group that was infected with LCMV Armstrong. Thus CD103 expression does not seem to be involved in controlling the antiviral CD8+ T-cell response during chronic LCMV infection.
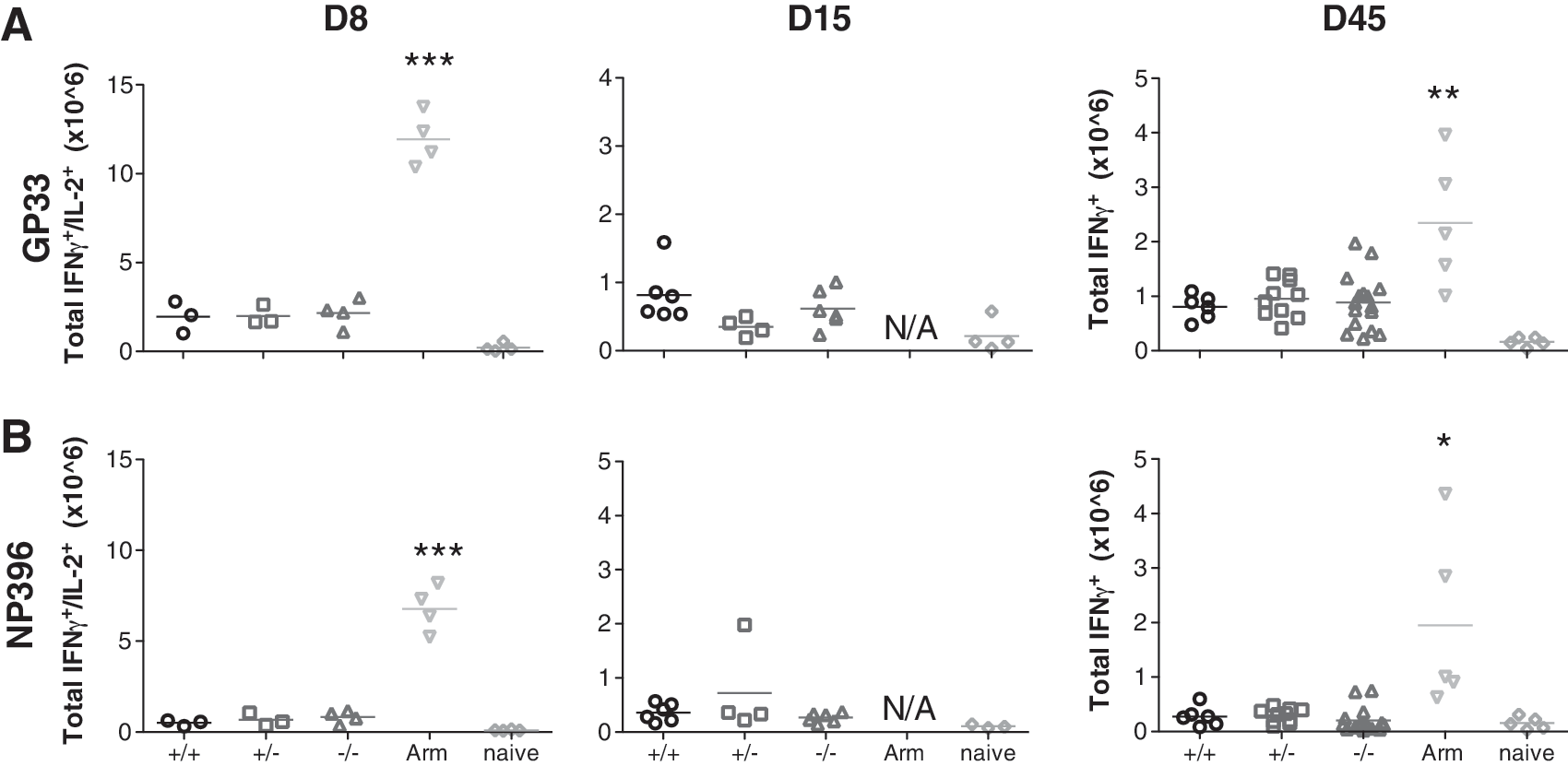
No alteration in functionally impaired CD8+ T cells was seen in the absence of CD103. CD103+/+, CD103+/−, and CD103−/− mice were infected with 2 × 106 PFU LCMV cl13 intravenously, whereas wild-type controls were infected with LCMV Armstrong (Arm). Spleens were collected at 8, 15, and >45 days after infection and cultured with the LCMV MHC-I peptides GP33 (
Discussion
In this study, we showed that the integrin CD103 plays a minimal role in functional impairment and exhaustion of T cells during chronic infection with LCMV cl13. Continuous replication depends on the generation of infectious LCMV virions despite ongoing antiviral immunity, which results in constant antigenic stimulation of lymphocytes, causing their gradual decline and thus exhaustion. Upon LCMV cl13 infection, CD103 levels considerably increased with time on CD4+CD25+ T cells, while they were transiently reduced on CD8+ T cells. CD103 upregulation on CD4+CD25+ T cells was long lasting and viral strain-specific, since it occurred only after cl13 and not Armstrong infection, correlating with disease progression. In contrast, CD103 levels on CD8+ T cells declined during the first 2 wk p.i. in spleen, mLN, and blood, regardless of the viral strain. In the mLN, however, CD103 expression levels were reduced much more profoundly in chronically-infected than in acutely-infected mice. Interestingly, in the gut-associated lymphoid tissue of patients with an active CD8+ T-cell response during chronic HIV infection, a decreased expression of CD103 by CD8+ T cells was also observed, thus sharing some degree of similarity with the LCMV cl13 murine model of persistent infection (43). To date, it has not been determined whether decreased expression of integrins correlates with disease progression, although after LCMV cl13 infection the effector functions and memory conversion of T cells were found to be identical in the absence of endogenous CD103 expression.
The severity of T-cell dysfunction seen during chronic infection correlates directly with the level of infection, expression levels of inhibitory receptors on the effector T cells, APC function, production of immunoregulatory cytokines, CD4 help, and Tregs. Upregulation of inhibitory receptors such as PD-1 by exhausted CD8+ T cells is an important mechanism of T-cell dysfunction during chronic viral infections. The inhibitory receptor PD-1 regulates functional T-cell impairment after LCMV cl13 infection in mice. Notably, PD-1 blockade even during the chronic phase of infection, allows recovery of T-cell function, resulting in a marked reduction in viral load (44,45). Other inhibitory receptors, on the other hand, such as LAG-3 and CTLA-4 seem non-essential (46 –48). In our study, exhausted CD8+ T cells expressed low levels of CD103 and high levels of the negative regulatory receptor PD-1. In addition, cytotoxicity and the production of cytokines such as IL-2 and TNF were equally reduced or lost. The observed CD103 low expression levels on CD8+ T cells may explain why in-vivo ablation of this integrin did not rescue the cytokine production or alter the impaired status of virus-specific CD8+ T cells.
It is believed that continuous viral replication alters the immune system and also causes tissue damage, which can be observed by the gradual weight loss in the affected host. Weight loss as a cause of LCMV cl13 infection was similar in the absence of endogenous CD103 expression. LCMV persists in specific cell types during chronic infection, particularly in the spleen, the liver and primarily the kidney, partially due to its tropism for fibroblastic reticular cells and dendritic cells (DCs) (49,50). The LCMV titers in the liver and kidney in wild-type and CD103−/− mice were almost identical. The virus was cleared somewhat faster from the kidneys of CD103 wild-type mice, since on day 15 p.i. higher virus titers were detected in the CD103−/− mice. This observation suggests that the kidney tissue microenvironment and some virus-dependent local inflammatory signals were transiently affected by the absence of CD103 expression.
CD103 is expressed not only by CD8+ T cells, but also by subsets of CD4+ T cells and some DCs residing in the gut wall (42,51,52). DCs, the key regulators of immune responses, play important roles in antiviral immune responses. In previous studies we showed that IL-10 secretion by a population of DCs (CD11c+CD8α) is implicated in preventing viral clearance, thus enabling viral persistence. Therapeutic IL-10R blockade broke the cycle of IL-10–mediated immune suppression and enhanced antiviral responses, thereby resolving infection in persistently infected mice (31,32). In addition to IL-10, other regulatory cytokines such as TGF-β1 have an impact during persistent viral infections through undefined mechanisms (46). TGF-β1, a cytokine that is highly expressed at the mucosal tissues and sites of inflammation, positively regulates the expression of αE integrin (CD103) (53). The CD103+ DCs from mLN express TGF-β1 and Aldh1a2, an enzyme that converts vitamin A to retinoic acid (RA), and the combination of TGF-β and RA effectively convert naïve T cells into Foxp3+ Tregs (54,55).
Tregs can paradoxically negatively or positively regulate effective immunity during acute infections, whereas in most cases Tregs seem to be involved in modulating effector T-cell recruitment to infected tissues (10,12,56). Treg deregulation has been reported to be present in many examples of persistent viral infections, although it is unclear if the altered Treg function is a cause or effect of viral persistence in the host. Recently it has been shown that CD103 was also expressed at the surface of natural CD4+CD25+ T cells from lymphoid organs, ∼30% of which express this molecule. This allowed the identification of subsets of Tregs without clearly defined properties. Importantly, CD103 expression was found to be critical for the suppression of mucosal inflammation, and furthermore is required for Treg retention in the skin after Leishmania major chronic infection (17). CD4+CD25+ Treg number and frequency were not significantly altered in the absence of CD103 expression during the course of a chronic infection. On the other hand, strong upregulation of CD103 on the surface of Tregs upon LCMV cl13 but not Armstrong infection was observed, which correlated with viral persistence. This suggests that even though CD103 is continuously upregulated on Tregs during chronic states, it does not significantly contribute to T-cell exhaustion.
Acknowledgments
We would like to thank Priscilla Colby for excellent administrative assistance. This work was supported by the Brehm coalition and by funds from U01DK. Georgia Fousteri is an American Heart Association scholar awardee.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.