
Abstract
Immunoglobulins in secretions play a critical role in protection at mucosal surfaces. We examined the generation of viral-specific IgG and IgA in plasma and mucosal secretions of mice following systemic or mucosal immunization with lymphocytic choriomeningitis virus (LCMV), a widely used experimental model of viral infection. While there are early differences in humoral responses depending on the route of viral entry, we show that both routes generate comparably robust viral-specific IgG in plasma, vaginal, lung, and nasal secretions of immune mice. In contrast, LCMV elicited poor viral-specific IgA responses. Mice that were infected IN showed elevated viral-specific IgA in nasal and lung washes compared to IP-infected mice; however, LCMV-specific IgG overwhelmingly contributed to the humoral response in all mucosal secretions examined. Thus similarly to HIV-1, and several other mucosally-encountered microbial infections, these data suggest that LCMV infection fails to induce vigorous viral-specific IgA responses.
Introduction
Experimental lymphocytic choriomeningitis virus (LCMV) was first described in 1934 following isolation from a female African-American patient in St. Louis, Missouri (2). Later work identified LCMV as a natural pathogen of rodents, primarily transmitted by inhalation of aerosolized virus from infected excretions, as well as bites from infected animals (5). Experimentally, LCMV infection of mice has been used extensively to study virologic and immunologic factors in viral control and spread. LCMV elicits robust antiviral T- and B-cell responses following either systemic or mucosal infection (7,17,38). LCMV viral control is dependent on a functional CD8 T-cell compartment; however, the antiviral humoral response contributes to virus control (3,35).
We have previously shown that following both intranasal (IN) and intraperitoneal (IP) infection with LCMV, memory CD8 T cells were generated in the vaginal mucosa that were protective against vaginal pathogenic challenge (38). Our findings were consistent with several studies that demonstrated that systemic immunization with replicating vectors can successfully generate protective T-cell immunity in the genital tract and other mucosal sites (26,37,39). In contrast to studies of genital tract T-cell responses, it is well-established that IN infection generates superior humoral responses in the genital tract (22). However, few studies have examined LCMV-specific IgA responses, and no published studies have examined these responses in genital secretions (23,34).
In this study we investigated whether systemic or mucosal infection with LCMV generated antiviral humoral responses in mucosal secretions such as genital washes. We compared whether these responses differed depending on the route of LCMV infection, and if so, whether these differences persisted into the memory phase. Here we describe our results demonstrating that despite early differences in the magnitude of the response, LCMV infection elicited similar, robust viral-specific IgG in plasma and mucosal secretions following both routes of infection. In contrast, the IN route was superior at eliciting LCMV-specific IgA in nasal and lung washes of immune mice, but we did not observe significant viral-specific IgA responses in vaginal washes or plasma. In general, LCMV failed to elicit vigorous viral-specific IgA responses at all sites examined. Taken together, these results suggest that LCMV-specific IgA contributes minimally to the overall humoral response at mucosal surfaces.
Materials and Methods
Mice and virus
Female C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and P14.Thy1.1 mice were kindly provided by Dr. Rafi Ahmed at Emory University School of Medicine and bred in-house in accordance with the University of Tennessee IACUC guidelines. LCMV-Armstrong, an acute strain of LCMV that is cleared in 1–2 wk, was provided by Dr. Rafi Ahmed (1). We adoptively transferred ∼2.5 × 104 Thy1.1 P14 transgenic CD8 T cells specific for the LCMV glycoprotein GP33-41 (1 × 106 total splenocytes) IV into recipient mice, and 1 d later the mice were infected with 2 × 105 pfu LCMV-Armstrong IP or IN, and sacrificed at the indicated time points post-infection (p.i.) as previously described (38). Female C57BL/6 mice that did not receive transferred cells were also infected IP or IN with 2 × 105 pfu LCMV-Armstrong.
Sample collection and ELISA
Plasma and mucosal secretions from mice that were infected with LCMV IN or IP were obtained from individual mice. To obtain plasma, blood was collected by retro-orbital bleeding using heparinized capillary tubes, centrifuged at 12,000 × g for 30 min at 4°C, and plasma was collected and stored at −80°C until use. For collection of vaginal washes, 50 μL of PBS was flushed gently in and out of the vaginal tract of anesthetized mice and the washes were frozen at −80°C until use. Nasal washes and lung washes (bronchoalveolar lavage or BAL) were collected in 0.5 mL or 1.0 mL PBS, respectively, with 0.1% BSA and antibiotics. ELISA was used to determine LCMV-specific antibody titers as previously described (1). Viral-specific antibody titer is expressed as the reciprocal of the highest dilution showing an optical density at 492 nm twofold above background. For mucosal secretions, in order to increase detection sensitivity, the ELISA protocol was slightly modified. Samples were incubated on LCMV antigen-coated plates overnight at 4°C, washed, and then incubated with biotin-conjugated antibodies for 4 h. The plates were incubated with avidin-HRPO (Vector Laboratories, Burlingame, CA) for 2 h, washed, incubated with substrate for 30 min, and the enzyme reaction was terminated. LCMV viral titers were quantified by plaque assay of tissue homogenates on Vero cell monolayers as previously described (1).
Intracellular cytokine staining and ELISPOT
GP61-80-specific polyclonal CD4 T cells were detected ex vivo as previously described (24). Briefly, lymphocytes were prepared from spleens of mice days 44–102 PI, and 1 × 106 splenocytes were incubated 5 h at 37°C with 2 μg of GP61-80 peptide or no peptide controls, in the presence of brefeldin-A. The cell suspensions were stained with monoclonal antibodies to CD4 and intracellular cytokines (IFN-γ and IL-2) using BD Cytofix/CytoPerm Plus Kits (La Jolla, CA) according to the manufacturer's instructions. Numbers represent the percentage of gated CD4 or GP61-80-specific CD4 T cells. For ELISPOT analysis, LCMV-specific IgG, IgM, and IgA antibody-secreting cells (ASCs) were detected as previously described (8,35). Briefly, lymphocytes were prepared from spleens and bone marrow of infected mice, and serial dilutions of cell suspensions were incubated 5 h at 37°C on MultiScreen 96-well filtration plates (Millipore, Billerica, MA) coated with LCMV antigen. Biotinylated anti-Ig of interest was added to the plates and they were incubated 48 h at 4°C, followed by incubation with avidin D-HRP (Vector Laboratories) for 1 h, and developed with an AEC solution. The spots in each well were counted using an inverted microscope, and data are expressed as the frequency of ASCs/106 cells.
Results
Early differences in LCMV-specific humoral responses at day 8
We have previously analyzed antigen-specific CD8 T-cell responses in the vaginal tract and tissues comparing IP versus IN LCMV-Armstrong infection using P14 transgenic CD8 T cells specific for the GP33-41 peptide of the LCMV glycoprotein (38). This study revealed that both routes of immunization generated similar, protective Ag-specific CD8 T-cell responses in the vaginal tract, despite early quantitative and qualitative differences. We also collected plasma and vaginal washes from these mice to examine LCMV-specific antibody levels. As shown in Fig. 1, our analysis of samples at day 8 p.i. indicated that LCMV generated robust viral-specific IgM and IgG in plasma; however, LCMV-specific IgM, IgG, and IgA isotypes were consistently reduced in mice infected IN compared to mice infected IP (Fig. 1A and C). We could detect LCMV-specific IgG in the vaginal washes of most IP-infected mice at day 8, but the level was reduced in mice infected IN (Fig. 1B). In contrast, we observed no evidence of LCMV-specific IgA in plasma or vaginal washes following either route of infection at day 8 (Fig. 1A and B).
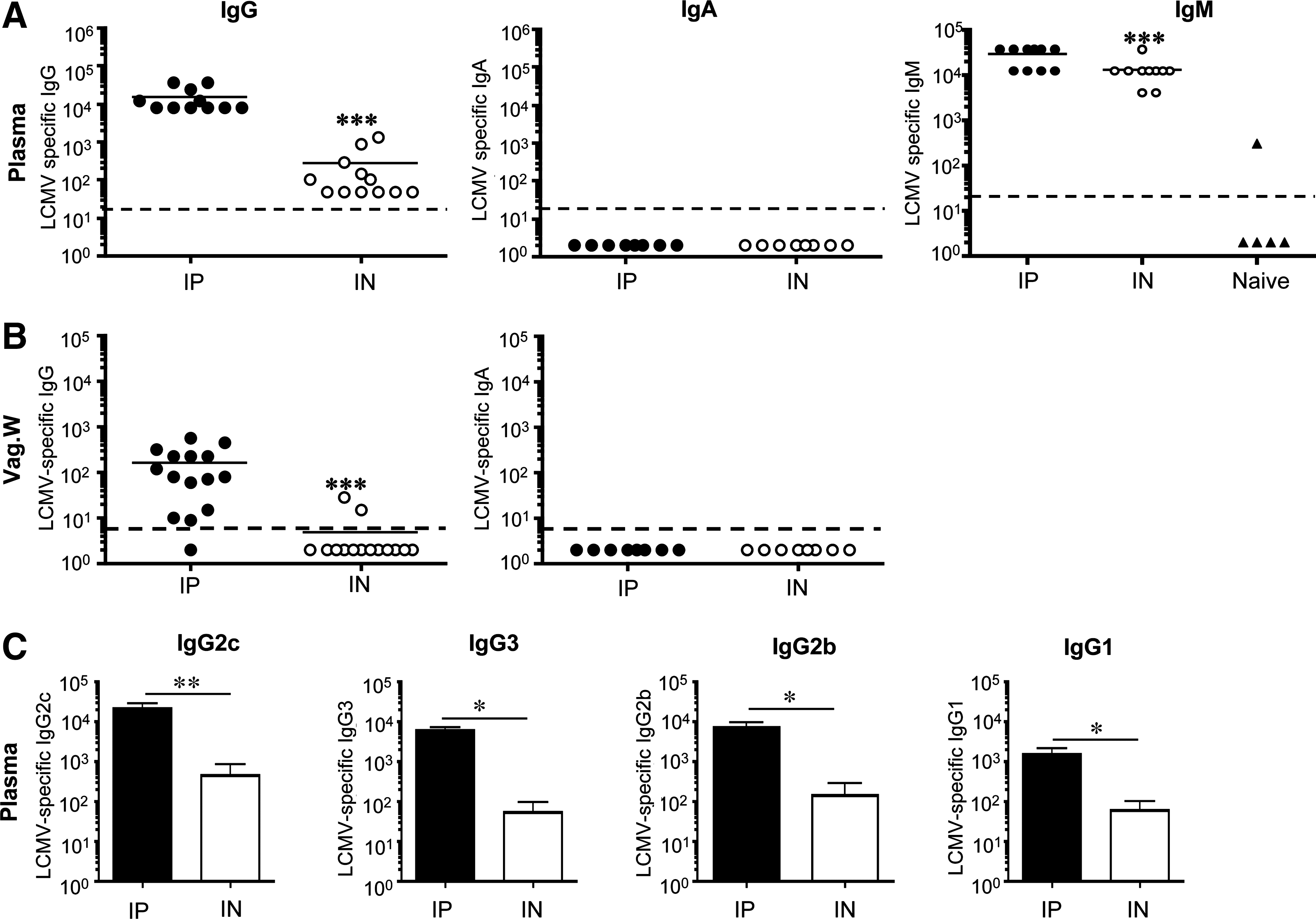
LCMV-specific humoral responses in plasma and vaginal washes at day 8 p.i. In each experiment groups of female mice were infected with 2 × 105 pfu LCMV-Armstong IP or IN, and LCMV-specific antibody responses were determined by ELISA at day 8 p.i. (
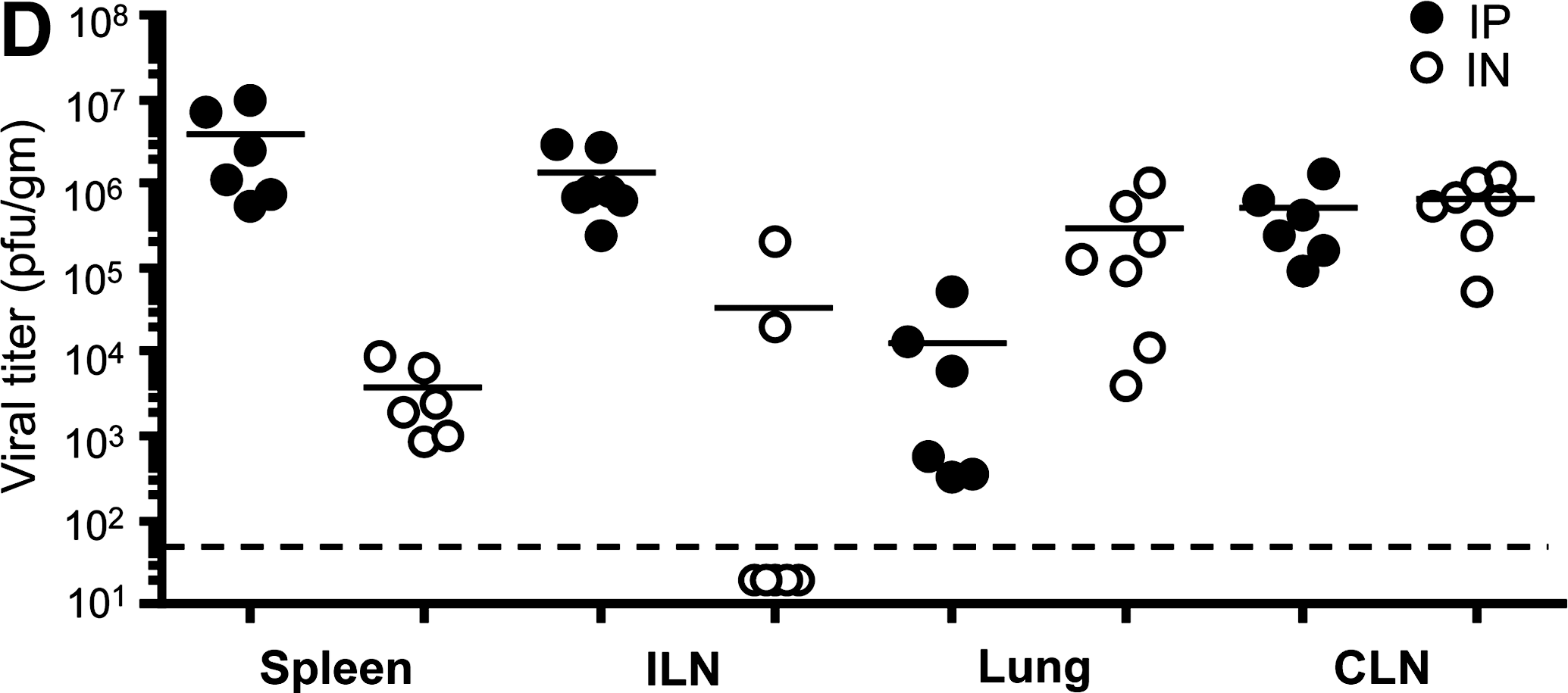
We next analyzed viral titers to compare viral replication in mucosal sites such as lung, versus systemic sites such as spleen, comparing the different routes of infection. Mice were infected with 2 × 105 pfu LCMV-Armstrong IP or IN, and viral titers in spleen, iliac lymph nodes (ILN), lung, and cervical lymph nodes (CLN) at day 3 p.i. were determined by plaque assay of tissue homogenates (Fig. 1D). As expected, we measured differences in the viral titer depending on the route of infection (29). Viral titers were higher in the spleens and ILN of IP-infected mice, higher in the lungs of IN infected mice, and similar in the CLN of both groups. These data suggest that early differences in LCMV viral replication, and consequently reduced viral antigen availability in some tissues, could potentially influence early differences in the induction of LCMV-specific responses. Notably, LCMV-specific IgG was not detectable in the vaginal washes of most IN-infected mice at day 8, and this may be due to low viral titers in the ILN of many of these mice (Fig. 1B and D).
Similar LCMV-specific humoral responses at memory
We next asked if these early differences in the humoral response persisted into memory. We analyzed LCMV-specific IgG and IgA humoral responses in the plasma and vaginal washes of immune mice. By day 15 and memory time-points (days 44–140), no differences in LCMV-specific IgG or IgG isotype levels in plasma were noted between the groups (Fig. 2A and E and data not shown). Significant levels of LCMV-specific IgG were present in the vaginal washes of both groups of mice, and we did not observe any statistically significant differences in the antibody titer (Fig. 2B). Moreover, analysis of total IgG in the vaginal washes of LCMV-immune mice versus naïve, uninfected mice, showed higher levels (∼10-fold) of total IgG for both IP and IN immune mice (Fig. 2C). In contrast, we detected little evidence of LCMV-specific IgA in the plasma or vaginal washes of IP- or IN-infected mice at any time point analyzed (Fig. 2A and B). Low levels of LCMV-specific IgA were detected in the vaginal washes of some mice, usually near the limit of detection for this assay (1/7). Analysis of total IgA in the vaginal washes of LCMV-immune mice was not significantly different from total IgA collected from the vaginal washes of naïve mice (Fig. 2D). We conclude from these data that LCMV infection fails to generate vigorous viral-specific IgA in the plasma or the mucosal surfaces of the genital tract, but does induce robust LCMV-specific IgG at these sites, regardless of the route of infection.
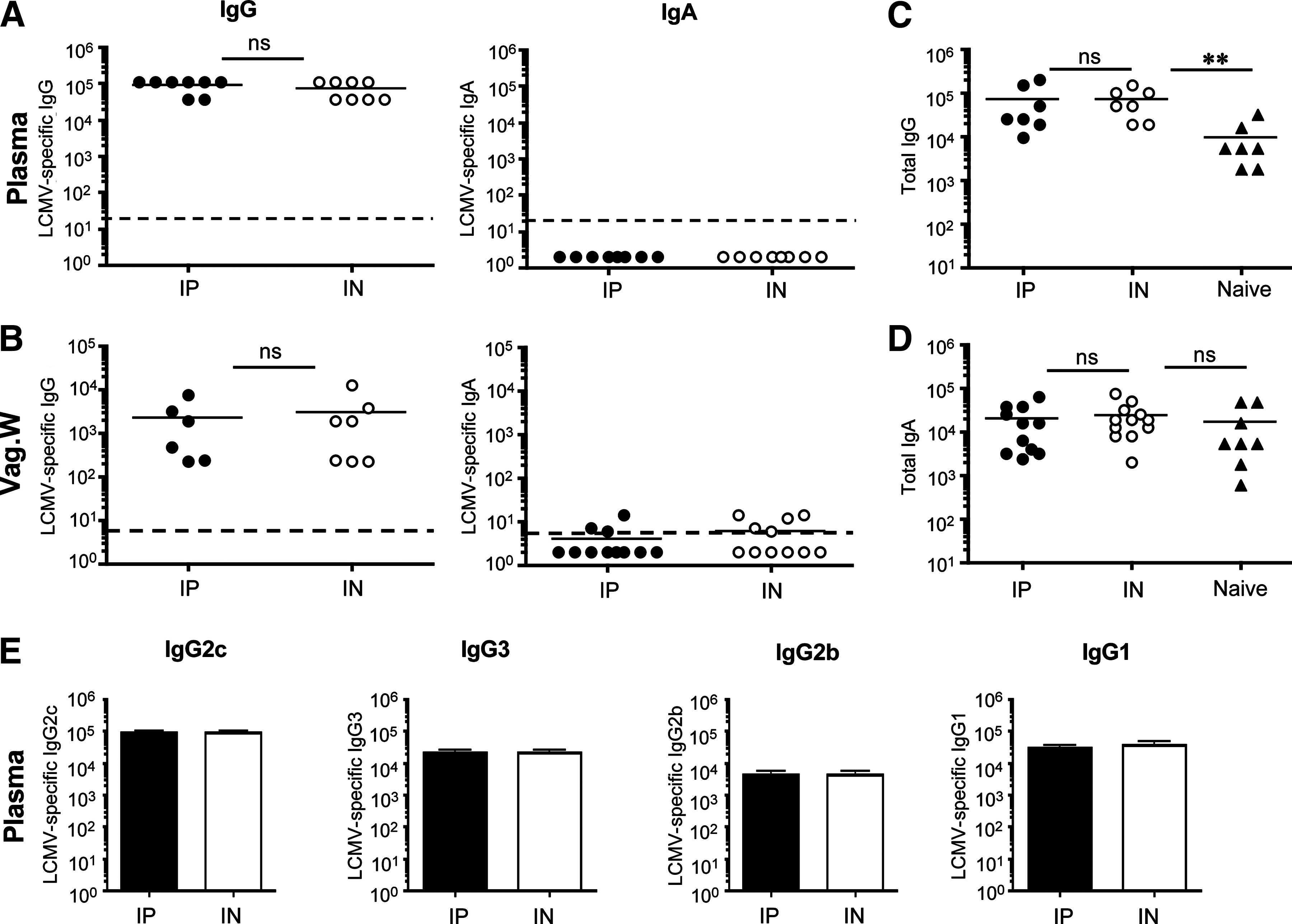
LCMV-specific humoral responses in plasma and vaginal washes at memory. Groups of female mice were infected with 2 × 105 pfu LCMV-Armstong IP or IN, and LCMV-specific antibody responses in vaginal washes were determined by ELISA at day 44 or 140 p.i. (memory includes data from days 44 and 140). (
Early differences in LCMV-specific humoral responses in mucosal and systemic compartments
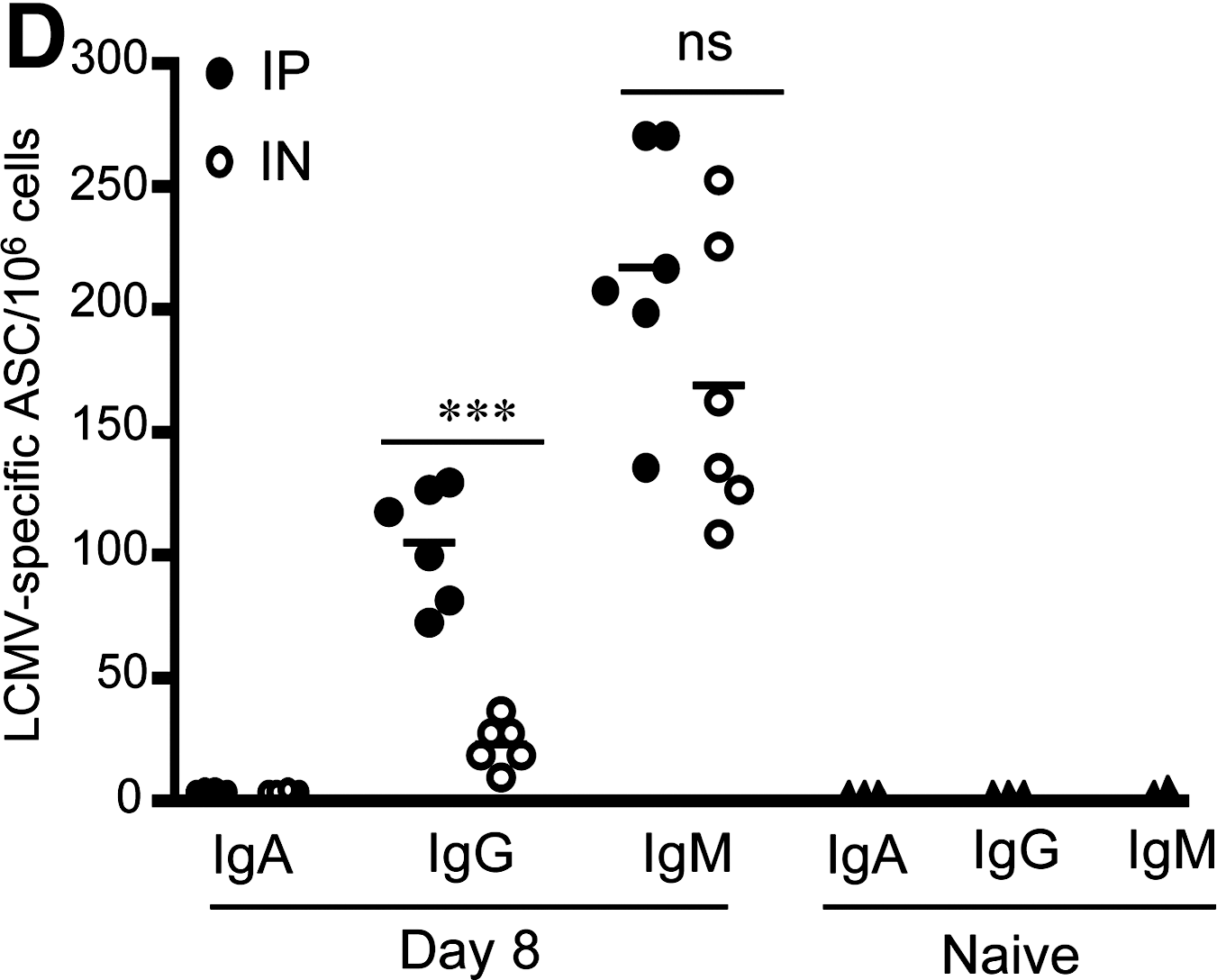
Our data thus far compared humoral responses in plasma and vaginal washes of infected mice that previously received transgenic LCMV-specific CD8 T cells (38). We considered whether addition of these T cells altered humoral responses in an unanticipated way. In addition, we sought to determine whether LCMV-specific IgA titers were elevated in mucosal secretions near the site of antigen exposure of IN-infected mice, such as nasal and lung washes, compared to distal sites, such as the genital tract. C57BL/6 mice were infected IP or IN with 2 × 105 pfu LCMV-Armstrong, and we obtained plasma, vaginal washes, nasal washes, and lung washes (BAL) at day 8. As we observed with the mice that received adoptive transfer of P14 transgenic CD8 T cells, there were early differences in the magnitude of LCMV-specific humoral responses depending on the route of infection (Fig. 3A and C). Consistently, mice that were infected IP showed elevated LCMV-specific IgG and IgM compared to mice infected IN at day 8 (Fig. 3A and C). Similarly to data presented in Fig. 1, LCMV-specific IgA levels were below the limit of detection for this assay in plasma and mucosal secretions at day 8 (Fig. 3B). Analysis of ASCs by ELISPOT analysis revealed little evidence of LCMV-specific IgA, but prominent LCMV-specific IgG and IgM ASCs consistent with the ELISA data, though there was more variability in the IgM responses (Fig. 3D). From these data we conclude that early after infection, the IP route is superior in generating a greater LCMV-specific antiviral humoral response, but both routes poorly induce LCMV-specific IgA.
LCMV-specific humoral responses at day 8. Groups of C57BL/6 female mice that did not receive transgenic T cells were infected with 2 × 105 pfu LCMV-Armstrong IP or IN, and LCMV-specific antibody responses in plasma, vaginal, lung, and nasal washes were determined by ELISA on day 8. (
LCMV-specific IgA responses in lung and nasal washes of IN immune mice
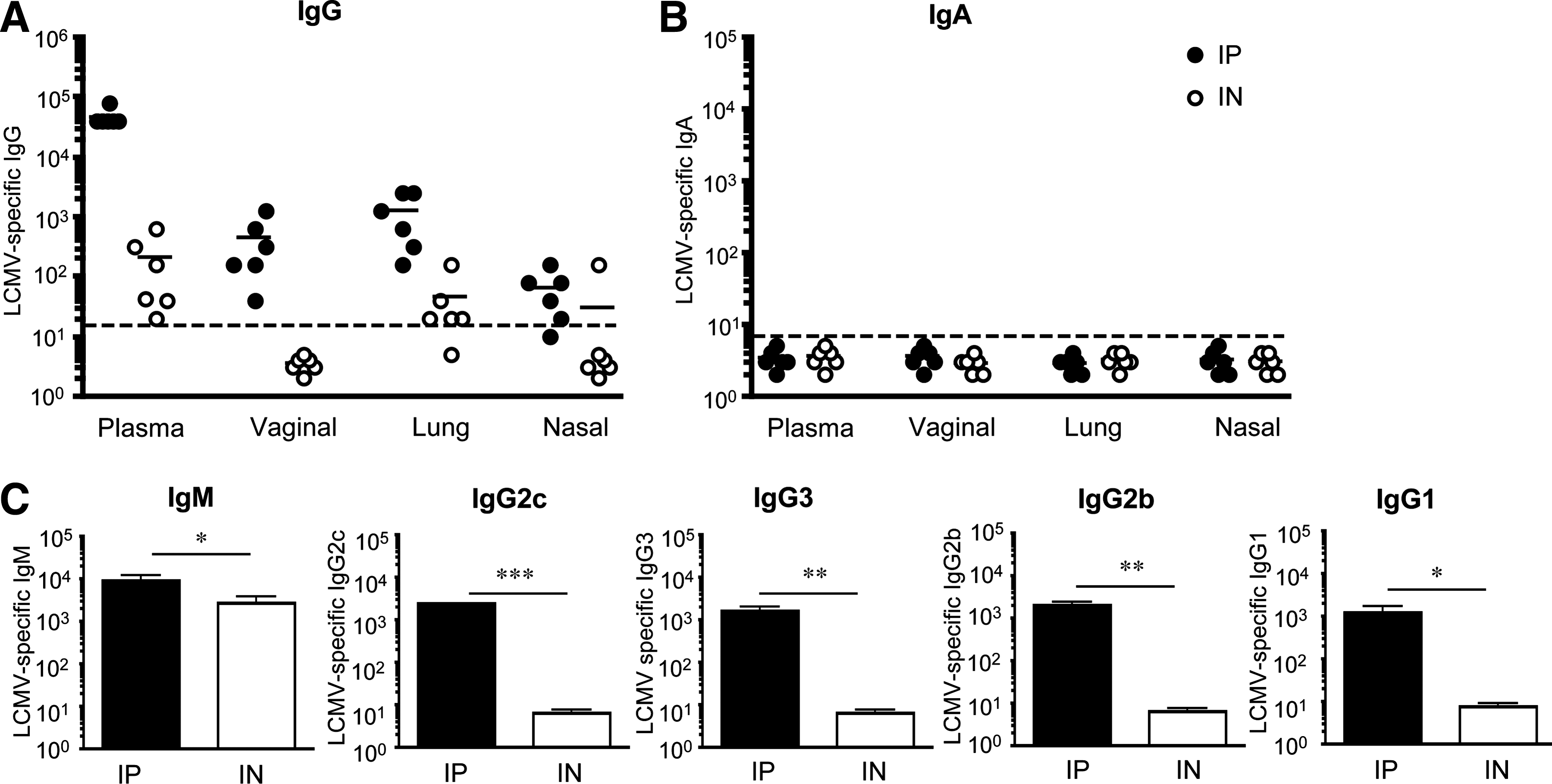
We then investigated whether these early differences in the magnitude of the humoral response persisted into the memory phase. Analysis of plasma and mucosal secretions of LCMV IP and IN immune mice (days 44–102) that did not receive transgenic P14 CD8 T cells, revealed robust LCMV-specific IgG antibody titers that were comparable in both groups (Fig. 4A). LCMV-specific IgG titers were highest in plasma and lowest in nasal washes. Additionally, while our analysis of LCMV-specific IgA responses in plasma and vaginal washes continued to be below the limit of detection, we consistently observed low LCMV-specific IgA responses in nasal and lung washes of most IN-infected mice, which were not detected in IP-infected mice (Fig. 4B). When we compared the proportion of LCMV-specific IgA of the total LCMV-specific antibody responses (LCMV-specific IgA/LCMV-specific IgG + IgA), in plasma, vaginal, lung, and nasal washes, we observed that the greatest contribution of LCMV-specific IgA to the humoral response occurred in the nasal washes, but only in IN-infected mice (Fig. 4C). At sites far away from the site of antigen exposure, the contribution of LCMV-specific IgA to the humoral response in the lung washes substantially dropped, as significantly more of the LCMV-specific humoral response was accounted for by LCMV-specific IgG at this site (Fig. 4C). In the vaginal washes and plasma, the contribution of LCMV-specific IgA to the total humoral response was negligible (Fig. 4C). Additionally, by memory time-points long-lived plasma cells had migrated from the spleen into the bone marrow, where LCMV-specific IgG ASCs overwhelmingly outnumbered LCMV-specific IgA ASCs, in both the IN and IP groups (Fig. 4D). For comparison, IN influenza immune mice showed similar frequencies of flu-specific IgG and IgA ASCs in the bone marrow (14).
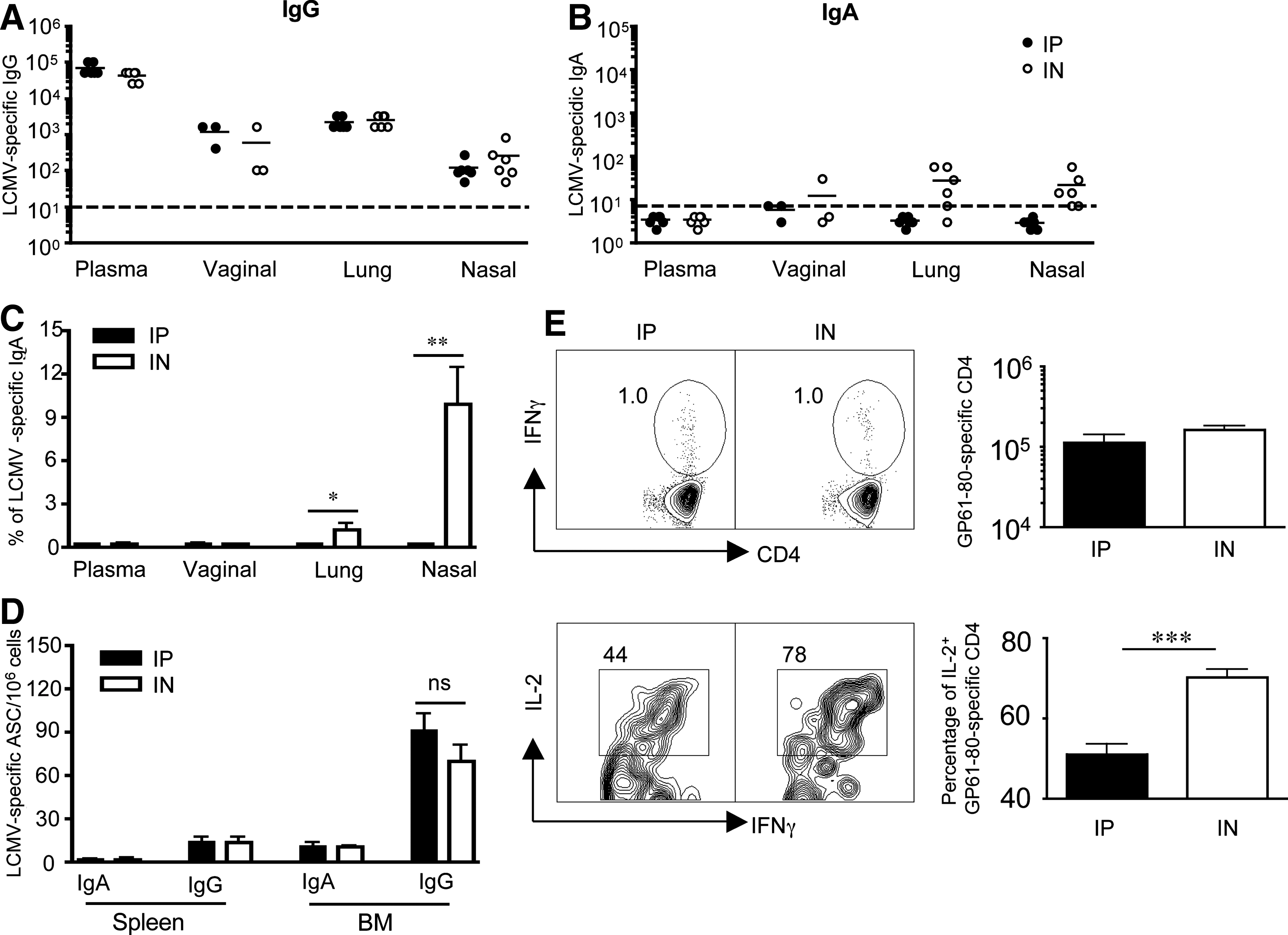
LCMV-specific humoral responses at memory. Groups of C57BL/6 female mice that did not receive transgenic T cells were infected with 2 × 105 pfu LCMV-Armstrong IP or IN, and LCMV-specific antibody responses in plasma, vaginal, lung, and nasal washes were determined by ELISA at memory (days 44 and 102 p.i.). (
Since IN infection generated LCMV-specific IgA responses in nasal and lung washes that were not detected in IP-infected mice, we hypothesized that the route of infection could program differences in the cytokine production of LCMV-specific CD4 T-helper cells following different routes of infection (27). We examined Ag-specific CD4 T-cell responses in both groups of immune mice. In support of this hypothesis, we observed that although both groups of mice showed similar numbers of GP61-80-specific CD4 T cells, a greater proportion of these Ag-specific CD4 T cells co-produced IL-2 (Fig. 4E). Additionally, we noted that GP61-80-specific CD4 T cells made more IFN-γ on a per-cell basis following the IN route of infection compared to the IP route of infection (mean fluorescence of IFN-γ+ was 260.7 ± 25.3 for IN versus 160.8 ± 16.3 for IP; p = 0.0058). Taken together, these data highlight the possibility that while the magnitude of the LCMV-specific immune response generated following IP infection can be comparable to IN infection at several sites, early differences in programming could result in qualitatively different immune responses that persist into memory.
Discussion
In summary, our data demonstrate that despite early differences in the magnitude of the antiviral humoral response, LCMV infection results in comparable, robust LCMV-specific IgG titers in the plasma and mucosal secretions of immune mice regardless of the route of infection. Taken together with our earlier study examining Ag-specific CD8 T-cell responses, these studies suggest that both systemic and mucosal LCMV infection are equally effective at generating robust memory T- and B-cell responses (16,19,26,37 –39). However, while these studies show that the magnitude of the T- and B-cell immune responses may be similar, and do not preclude protection against pathogenic challenge, they also show that initial route of infection, as well as tissue microenvironment, contribute to qualitative differences in the immune response in different tissues (38). These differences could potentially influence qualitative changes in the anamnestic responses at these sites under certain conditions that we were not able to discern in our studies.
We were surprised to discover that LCMV fails to generate vigorous viral-specific IgA responses, even when the virus was introduced via a mucosal route. While we were able to detect LCMV-specific IgA responses, particularly in the nasal washes, the site of antigen exposure of mice infected IN, these responses were characteristically weak compared to those of other mucosally encountered viral infections such as influenza. Influenza-specific IgA following IN immunization almost exclusively contributes to the total antiviral humoral response in nasal washes (>95% versus ∼10% in LCMV), and prominently contributes at other mucosal sites (15,44, and M.Y. Sangster, personal communication). Our results echo findings seen with other mucosally encountered pathogens, such as HIV-1, SIV, and murine gammaherpes virus (MHV-68), which have also been found to elicit poor viral-specific IgA responses (21,22,32,33).
There have been very few published studies examining LCMV-specific IgA responses. Our data are consistent with data presented in two previous studies examining LCMV-specific IgA. In one study, examining postnatal infection in human patients, 13/20 patients demonstrated low LCMV-specific IgA responses that were short-lived (34). In another study of LCMV infection in NMRI mice, researchers measured low numbers of LCMV-specific IgA ASCs in spleens, that could not be boosted upon re-infection, in contrast to LCMV-specific IgG (23). Taken together, all three studies suggest that LCMV infection of humans and mice elicits poor viral-specific IgA responses, but induces vigorous LCMV-specific IgG and Ag-specific T cells at mucosal sites. Potent antiviral IgA, as well as antiviral IgG, have been shown against another arenavirus in gut washes, Lassa fever virus, using an oral route of immunization with recombinant Salmonella and/or vaccinia vectors expressing Lassa virus nucleocapsid protein (9,30).
Several recently published studies have underscored the crucial role played by the production of cytokines by CD4 T follicular helper (TFH) cells in IgA class switching (10,41). Cytokines implicated in IgA class switching include IL-2, IL-4, IL-5, IL-6, IL-10, IL-21, and TGF-β (6,36). We speculate that differentiation of antigen-specific CD4 TFH cells following different routes of infection may skew cytokine secretion by TFH cells, and these responses may influence whether pathogens elicit robust pathogen-specific IgA. In support of this hypothesis, we noted that although both IP and IN LCMV infection generated similar numbers of Ag-specific CD4 T cells, a greater proportion of the Ag-specific CD4 T cells in IN-infected mice co-produced IL-2, and produced higher levels of IFN-γ, suggesting qualitative differences in the CD4 T-cell immune response (Fig. 4). Furthermore, recent work has shown that mucosal vaccination with recombinant LCMV-GP papillomavirus vectors expressing IL-2 can boost specific IgA responses at mucosal surfaces compared to vector only (11).
Some viruses directly interfere with antiviral immune responses. HIV-1 has recently been described to perturb antiviral responses of systemic and mucosal B cells using an immunosuppressive viral protein, Nef (42). Thus understanding how some viral pathogens potentially evolve mechanisms that interfere with the host's ability to elicit viral-specific IgA responses may have implications for efforts to boost humoral immunity at mucosal surfaces. Studies examining oral versus systemic immunization with LCMV showed that primary infection occurs in the gastric epithelium following oral (intragastric) infection, with dissemination of virus to tissues via lymphatic cells in a G-protein-coupled signaling-dependent manner (29,43). Consistent with these earlier observations, more recent studies suggest that LCMV preferentially targets DCs for infection and activation, with recombinant LCMV vectors producing potent cell-mediated responses, while the neutralizing antibody response is dependent on the surface glycoprotein expressed by the recombinant viral vector (4,12,28). Since DC-produced TGF-β1 appears to be a primary regulator of IgA production, another possibility for consideration is that targeted infection of DCs by LCMV interferes with the induction of LCMV-specific IgA responses (20,36,40). However, the relatively poor induction of neutralizing antibody responses following LCMV infection has also been shown to be an intrinsic feature of the envelope glycoprotein of LCMV, and it is also possible that poor immunogenicity of the glycoprotein plays a role in poor IgA induction (28). Future investigations to identify the mechanisms responsible for the poor induction of LCMV-specific IgA responses, may aid in the discovery of novel pathways to boost humoral immunity at mucosal sites.
Footnotes
Acknowledgments
We thank Shane Crotty, Mark Sangster, and Rafi Ahmed for helpful discussions and reagents. We thank Barry Rouse for critical comments on the manuscript. This work was supported by National Institutes of Health grant AI05771901, and University of Tennessee start-up funds to Thandi M. Onami.
Author Disclosure Statement
No competing financial interests exist.