
Abstract
Immune responses against hepatitis C virus (HCV) have been studied by numerous groups. However, details concerning the production of antibodies to antigenically variable epitopes remain to be elucidated. Since the sequences of the variable regions of several HCV proteins are different among the virus strains infecting patients, we decided to design peptide combinations that represent the theoretical maximum antigenic variation of each epitope to be used as capture antigens. We prepared six peptide mixtures (hypervariable epitope constructs; HECs) representing six different epitopes from structural and non-structural proteins of HCV from genotypes 1–6. Plasma from 300 HCV patients was tested to determine if their antibodies recognize the synthetic constructs. All the patients were chronically infected with diverse HCV genotypes and did not receive antiviral treatment. Antibodies to one or more of the HECs were detected in all of the HCV-infected individuals. Immunogenicity of the HCV HECs was also evaluated in outbred and inbred mice. Strong HEC-specific antibodies were produced, and cellular responses were also induced that were Th-1 rather than Th-2. Our results show that HCV HECs are both antigens that can be used to detect the broad cross-reactivity of antibodies from HCV-infected patients, and strong immunogens that can induce antigen-specific humoral and cellular immune responses in mice.
Introduction
HCV was classified as non-A, non-B hepatitis (NANBH) until it was identified in 1989 by isolating its RNA genomic sequence from experimental chimpanzee plasma using random primers (11). Since identified as the causative agent, HCV is now recognized as one of the most serious public health problems, infecting an estimated 3% of the world's population (about 170 million people worldwide). HCV is a major cause of chronic liver infection that can lead to cirrhosis and hepatocellular carcinoma (HCC) (32). HCV is an enveloped, single-stranded positive-sense RNA virus that belongs to the Flaviviridae family. HCV encodes a single open reading frame (ORF) of about 9600 bp nucleotides in length, flanked by a 5′ and a 3′ untranslated region (UTR). The ORF encodes a polyprotein precursor that is processed post-translationally by cellular and viral proteases to produce structural and nonstructural proteins, respectively. The structural proteins consist of core, two envelope proteins called E1 and E2, and the non-structural proteins NS2, NS3, NS4A, NS4B, NS5A, and NS5B (4).
Based on sequence variation, HCV has been classified into six major genotypes that differ by approximately 30% from one another (24,26,32). Within each genotype of HCV, there are several subtypes with nucleotide differences of approximately 20–25% (24,32). Multiple viral variants present in the blood of a given individual (quasispecies) can differ by as much as 10% (24). Epitope variability has been observed in the structural E1 and E2 envelope glycoproteins, as well as in the non-structural NS3, NS4, and NS5 proteins (4,10,36 –38). Hypervariability exists mostly in the amino-terminal portion of the E2 protein, in a region named the first hypervariable region (HVR1) (17,26,34). HVR1 is the major neutralizing epitope of HCV, and consists of 27 amino acids (26). A second hypervariable region (HVR2), in the carboxyl-terminal region of the E2 glycoprotein, consists of nine amino acids (34).
Hypervariable epitope constructs (HEC) are synthetic peptide mixtures that contain multiple variants of a given epitope based on hypervariable regions of viruses that mutate their genomic sequences frequently to evade immune responses. The method for designing the HEC based on these regions has been described elsewhere (2,3,8,9,22,23). Our previous work demonstrated that HECs based on hypervariable regions of simian immunodeficiency virus (SIV) or human immunodeficiency virus (HIV) induce broadly reactive humoral as well as T-helper cell responses in rodents and non-human primates (2,3,8,22,23). Here, we apply the same principle to develop immunogens that could be part of a diagnostic test or vaccine candidate against HCV.
HCV HECs are composed of six antigenic variable epitopes representing the six major genotypes and their subtypes circulating in the HCV-infected population. Over 300 HCV protein sequences were obtained from the Genbank database. Design of the HEC was based on analysis of amino acids present among antigenically variable sequences of HCV from subtypes 1a, 1b, 2a, 2b, 3, and 4-6, with the number of sequences from each HCV subtype representing approximately 30%, 25%, 15%, 15%, 10%, and 5% of the total number of sequences, respectively. Two epitopes are from the E2 envelope glycoprotein (HVR1 and HVR2 HEC), two epitopes are from the NS3 protein (NS3-1 and NS3-2 HEC), and two epitopes are from the NS4 protein (NS4-1 and NS4-2 HEC). HCV HECs were used as capture antigens to detect antibodies in patients with HCV chronic infection, and as immunogens for examination of induction of specific immune responses in mice.
Single-sequence peptides (termed analogs) derived from the HVR1 and HVR2 regions of the E2 envelope glycoprotein were prepared to determine if antibodies from mice immunized with HCV HEC could also recognize each region. Each HVR1 and HVR2 analog represents a specific epitope from a single viral isolate of each genotype. HVR1 and HVR2 analogs contain sequences from genotypes 1 to 6 (1a, 1b, 2a, 2b, 2e/f, 3a, 4a, 5a, and 6a).
Numerous groups have been studying the immune responses against HCV, but the production and breadth of reactivity of antibodies to antigenically variable epitopes representing diverse genotypes and subtypes remain to be elucidated. In the present study, we assessed if HECs are recognized by antibodies from individuals infected with diverse genotypes and subtypes of HCV (1a, 1b, 1a/b, 2a, 2b, 2a/c, and 3a). Plasma from two different groups of patients with chronic HCV infection was tested as follows: (1) group 1 consisted of 228 HCV patient samples that were tested for antibody binding to HVR1 and HVR2 HECs, and (2) group 2 consisted of 49 HCV patient samples tested for antibody binding to the six HCV HECs. We also tested the antigenicity of HCV HEC by assessing induction of humoral and cellular immune responses in mice. Different combinations of HCV HECs were used to immunize mice, and HEC-specific antibody responses, lymphocyte proliferation, and cytokine production were determined.
Materials and Methods
Plasma samples
Archived patient plasma samples were obtained from the AIDS Research Program at the Ponce School of Medicine in Puerto Rico and confirmed positive for HCV by RT-PCR. HCV genotypes were determined by line probe assay (Inno-LiPA HCV II; Innogenetics, Ghent, Belgium). All the plasma samples were from patients chronically infected with different genotypes of HCV (1a, 1b, 1a/b, 2a, 2b, 2a/c, and 3a), and had never received HCV antiviral treatment (i.e., IFN-α and ribavirin). All the plasma samples were first screened by the Abbott HCV EIA 2.0 test (Abbott Laboratories, Abbott Park, IL), that utilizes recombinant antigens c100-3, HC-31, and HC-34. HCV viral loads were determined by the Amplicor HCV Monitor procedure (Roche Diagnostics Corp., Indianapolis, IN).
HEC synthesis
Peptides representing six antigenic variable epitopes of six HCV genotypes and their subtypes (1a, 1b, 2a, 2b, 3, and 4-6), including the hypervariable regions of E2, NS3, and NS4, were synthesized. The sequences of HCV HECs are shown in Table 1. The procedure of HEC synthesis has been previously described (8–9). In brief, all HCV HECs were synthesized by 9-fluorenylmethyloxycarbonyl (Fmoc) chemistry, a solid-phase peptide synthesis method utilizing high-capacity (0.7 mmol/g) Knorr resin (Advanced ChemTech, Louisville, KY) (8). Synthesis was performed using an automatic peptide synthesizer (PS-3; Rainin Protein Technologies, Woburn, MA). The appropriate molar amounts of amino acids were added based on frequencies calculated for a given position. The final peptides were cleaved and deprotected by addition of trifluoroacetic acid with scavengers. The peptide mixture was suspended, lyophilized, and dialyzed in distilled water at least three times to remove acid residues from cleaved peptide. Amino acid analysis was performed on all HECs to ensure that they had the appropriate amino acid content.
A bar in the HEC sequence indicates an identical amino acid.
Positions where two amino acids were added are shown, as well as the individual sequence variants expected in each of the final constructs.
Abbreviations: HEC, hypervariable epitope construct; HCV, hepatitis C virus; HVR, hypervariable region.
HVR1 and HVR2 HEC analog peptide synthesis
Eleven single-sequence peptides derived from both the HVR1 and HVR2 regions of the E2 protein from a single genotype of each viral isolate were synthesized and used as capture antigen on ELISA plates. We designated these single-sequence peptides as HVR1 analog and HVR2 analog to distinguish these from the HECs, and the sequences are shown in Table 2. Each peptide represents epitopes from different genotypes and subtypes of HCV (1a, 1b, 2a, 2b, 2e/f, 3a, 4a, 5a, and 6a). Two peptides from the same genotype (i.e., 1a and 1b) came from different viral isolates with different amino acid sequences. The synthesis procedure was the same as that described above.
Abbreviations: HEC, hypervariable epitope construct; HCV, hepatitis C virus.
HCV viral purification from patient plasma
HCV viral particles were purified using an HCV purification kit (BioVintage, San Diego, CA), according to the manufacturer's instructions. Briefly, HCV viral particles were released from HCV-infected patient plasma by three freeze/thaw cycles. Supernatants were applied to mini-spin filter columns and centrifuged at 2000 g for 5 min. The columns were washed with wash buffer, followed by collection of HCV viral particles from the columns with elution buffer. Viral protein concentration was measured by Bradford assay.
Enzyme-linked immunosorbent assay (ELISA)
Binding of antibodies from HCV-infected patients to HCV HEC
ELISA was performed to quantitate antibodies present in patient plasma that bound to HCV HEC, as previously described (9). Briefly, all the HCV patient plasma samples were heat-inactivated at 56°C for 30 min before being used in assays. Flat-bottomed microtiter plates (Corning Costar 9018) were coated with HEC individually and held at 37°C overnight at a concentration of 5 μg/well in triplicate. Non-fat milk (10%) was added and the plates were kept for 2 h at 37°C to block non-specific binding, then the plates were washed with buffer before HCV patient plasma samples were added and incubated for 2 h at 37°C. Plasma from HCV-infected patients and three control human plasma samples negative for HBsAg, HCV, HIV-1, and HTLV-1 were tested at 1:100, 1:500, 1:1000, 1:5000, and 1:10,000 dilutions. Two of the control plasma samples were obtained from the Sacramento Blood Bank, and the third control was commercially-available normal human plasma (Biocell Laboratories, Inc., Rancho Dominguez, CA). Following incubation with primary antibody, the plates were washed and alkaline-phosphatase-labeled secondary antibody (goat anti-human IgG AP; Southern Biotechnology Associates, Birmingham, AL) was added and incubated for 1 h at 37°C. After the plates were washed, p-nitrophenyl phosphate substrate (KPL) was added and incubated for 2 h at room temperature. Optical density (OD) was measured spectrophotometrically at 405 nm with an automatic plate reader (VERSAmax; Molecular Devices, Sunnyvale, CA) according to the manufacturer's instructions (SOFTmax PRO; Molecular Devices). End-point antibody titer was defined as the greatest dilution of sample that maintained an OD at least twice that of the average of control plasma tested at the same dilution. The mean standard deviation of the OD readings was calculated.
Immunogenicity of HCV HEC in mice
ELISA for mouse sera was performed similarly to that described above. HCV HEC was coated individually on the plate at a concentration of 5 μg/well in triplicate. Mice sera were added as primary antibody, followed by alkaline-phosphatase-labeled secondary antibody (goat anti-mouse IgG AP, Southern Biotechnology Associates). The rest of the procedure was performed as described above.
Antibody response of mice immunized with HCV HEC to HCV viral particles purified from HCV-infected patient plasma
ELISA plates were coated with 10 μg/well of HCV viral particles purified from plasma of HCV-infected patients. The rest of the procedure was performed as described above.
Mouse immunization
BALB/c (4 mice per group) and CFW (6 mice per group) mice were immunized with a single HCV HEC and in combination. For each immunization, 100 μg of the peptides in 50 μL of sterile PBS was mixed at a 1:1 ratio with the adjuvant Montanide ISA-51 in a final volume of 100 μL. Immunizations were administered a total of four times at monthly intervals. All immunizations were delivered subcutaneously at the base of the tail.
Collection of spleens and lymph nodes from mice
Mice were euthanized 7 days after the final immunization. Spleen and lymph nodes (inguinal, axillary, and mesentery lymph nodes) were collected and pushed through a 70-μm cell strainer (BD Falcon; BD Biosciences Pharmingen, Franklin Lakes, NJ) using sterilized phosphate Dulbecco's saline solution with 2% bovine calf serum (HyClone; Thermo Fischer Scientific, Rochester, NY), and lymphocytes were isolated and separated from red blood cells on Ficoll-Hypaque (ICN Biomedicals Inc., Costa Mesa, CA). Cells were counted by the trypan blue exclusion method and resuspended in RPMI 1640 (HyClone) supplemented with 10% bovine calf serum and 1% antibiotic-antimycotic (Invitrogen Corp., Carlsbad, CA).
Lymphoproliferation assay
Determination of antigen-specific lymphocyte proliferation was performed by the thymidine incorporation method. Lymphocytes from spleens and lymph nodes (2 × 105 cells/well) were seeded on round-bottom 96-well tissue culture plates in triplicate in the presence of HCV HEC individually at a concentration of 5 μg/mL. As negative control, an HEC representing an epitope of the envelope glycoprotein of SIV and antigenically unrelated to HCV was used at the same concentration. The cells were incubated with the HCV HEC at 37°C in 5% CO2 for 3 d. Then 1 μCi of [3H]thymidine (Amersham Biosciences, Piscataway, NJ) was then added to each well in 50 μL of RPMI 1640 supplemented with 10% bovine calf serum and 1% antibiotic-antimycotic. Following a 12- to 14-h incubation period, the plates were harvested using a PHD cell harvester, and the amount of incorporated tritiated thymidine was measured with a Beckman LS 6000IC scintillation counter. The results are expressed as counts per minute (cpm).
Cytokine ELISA
Secretion of IFN-γ, IL-2, and IL-4 by HEC-specific lymphocytes was determined by ELISA. The lymphocytes were seeded the same way as for the lymphoproliferation assay method described above, except for the number of cells (5 × 105 cells/well). The cells were incubated at 37°C in 5% CO2 for 2 d. Supernatants were harvested after 48 h of stimulation with a 5-μg/mL concentration of individual HEC. Antibody pairs and standards were purchased from eBioscience (San Diego, CA). Cytokine capturing antibody-coated ELISA plates were blocked with 5% bovine calf serum (BCS)/PBS before supernatants and standard dilutions were added at 100 μL per well. Following washing six times with PBS in 0.05% Tween-20, biotinylated detecting antibodies were added at 100 μL per well in 1% BCS/PBS. After the plates were washed, streptadivin-conjugated horseradish peroxidase (Zymed Laboratories, Carlsbad, CA) was added at 100 μL per well and incubated for 30 min. Following a final wash, TMB Microwell peroxidase substrate (1-component; KPL Inc., Gaithersburg, MD) was added. Stop solution (KPL) was added after 30 min of incubation, and the plates were read at 450 nm with an automatic plate reader (VERSAmax; Molecular Devices), according to the manufacturer's instructions.
Statistical analysis
The difference between the groups was calculated by using ANOVA for normally distributed variables (GraphPad InStat version 3.0; GraphPad Software, San Diego, CA).
Results
Binding of patient antibodies to HVR1 and HVR2 HECs
Two well-known hypervariable regions are present in the E2 envelope protein (6,17). The HVR1 and HVR2 HECs represent 32 (HVR1) and 16 (HVR2) different variants based on those two regions. To determine if HCV HVR1 and HVR2 HECs were recognized by antibodies from HCV-infected patients, 228 plasma samples (group 1) from patients infected with different genotypes and subtypes of HCV (1a, 1b, 2a, 2b, and 3a) were screened by ELISA. The HCV patient plasma collection included 104 samples from patients infected with genotype 1a, 76 samples with genotype 1b, 20 samples with genotype 2a, 12 samples with genotype 2b, and 16 samples with genotype 3a. The results demonstrate that more than 80% of plasma from 228 patients contained antibodies that bound to HCV HVR1 (84%) and HVR2 (80%) HECs (Table 3). The results also showed that antibody reactivity to HCV HEC was not restricted to one specific genotype, but was broad, including all the HCV genotypes tested (Fig. 1). Interestingly, the end-point antibody titers to HVR1 HEC from patients with different genotypes and subtypes of HCV tested was higher than that to HVR2 HEC, which suggests that more antibodies specific to the HVR1 region were produced than to the HVR2 region when patients were chronically infected with HCV. It also suggests that a stronger cross-reactive antibody response was induced against the sequence variation of the HVR1 region than to the HVR2 region in patients with persistent chronic HCV infection.
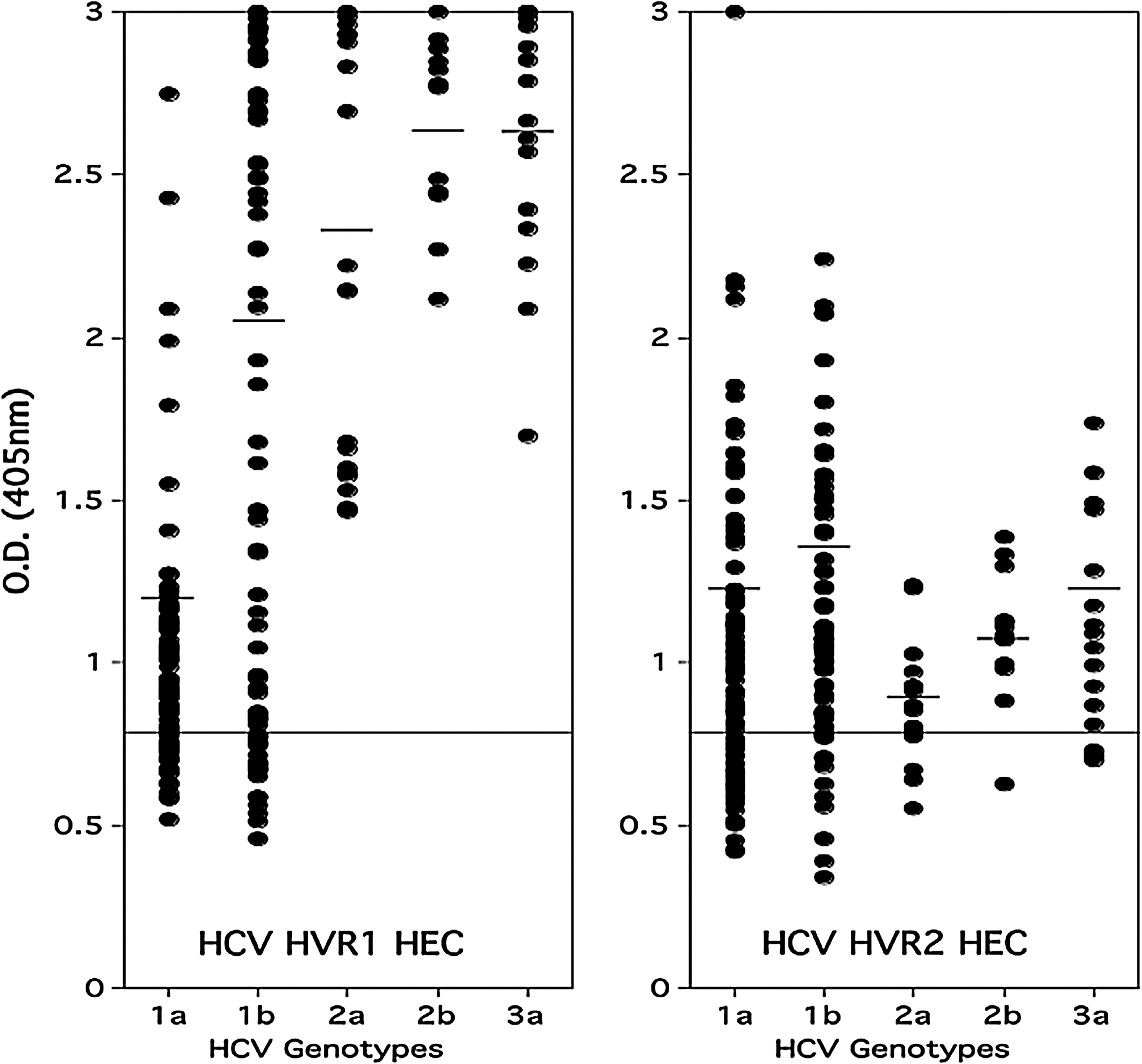
Relative antibody titers to the HVR1 and HVR2 HECs in plasma of HCV-positive patients (group 1). Dot plot showing relative OD values obtained when individual plasma samples from HCV patients were tested for antibodies to HVR1 and HVR2 HECs. All sera were diluted 1:100. The samples were considered positive if their optical density at 405 nm was more than twice that of the negative sera. The horizontal line indicates the OD value that is twice the average of four HCV-negative serum samples. The sera did not react with a negative control HEC based on HIV-1 (data not shown).
Abbreviations: HEC, hypervariable epitope construct; HCV, hepatitis C virus; HVR, hypervariable region.
Binding of patient antibodies to six HCV HECs
Based on the results obtained with patient group 1, we decided to test a second group of patients (group 2) for antibody reactivity to NS3-1, NS3-2, and NS4-1, NS4-2 HCV HECs, in addition to HVR1 and HVR2 HECs. Antibody reactivity to each HEC and the percentages of patients that responded to each HEC are shown in Fig. 2 and Table 4, respectively. We determined the end-point titer of each plasma sample by ELISA up to a 1:10,000 antibody dilution. Group 2 consisted of plasma from 50 randomly-selected HCV-positive patients infected with diverse genotypes and subtypes of HCV (1a, 1b, 1a/b, 2b, 2a/c, 3a, and 4c/d). The HCV genotype and subtype distribution were 20 (1a), 7 (1b), 2 (1a/b), 9 (unknown subtype of genotype 1), 3 (2b), 2 (2a/c), 6 (3a), and 1 (4c/d). Viremia in circulating blood was determined by RT-PCR. The viremia of all patients selected was at least 315,000 IU/mL. Viremia of 32 out of the 50 patients was more than 850,000 IU/mL, and more than 500,000 IU/mL in 11 patients.
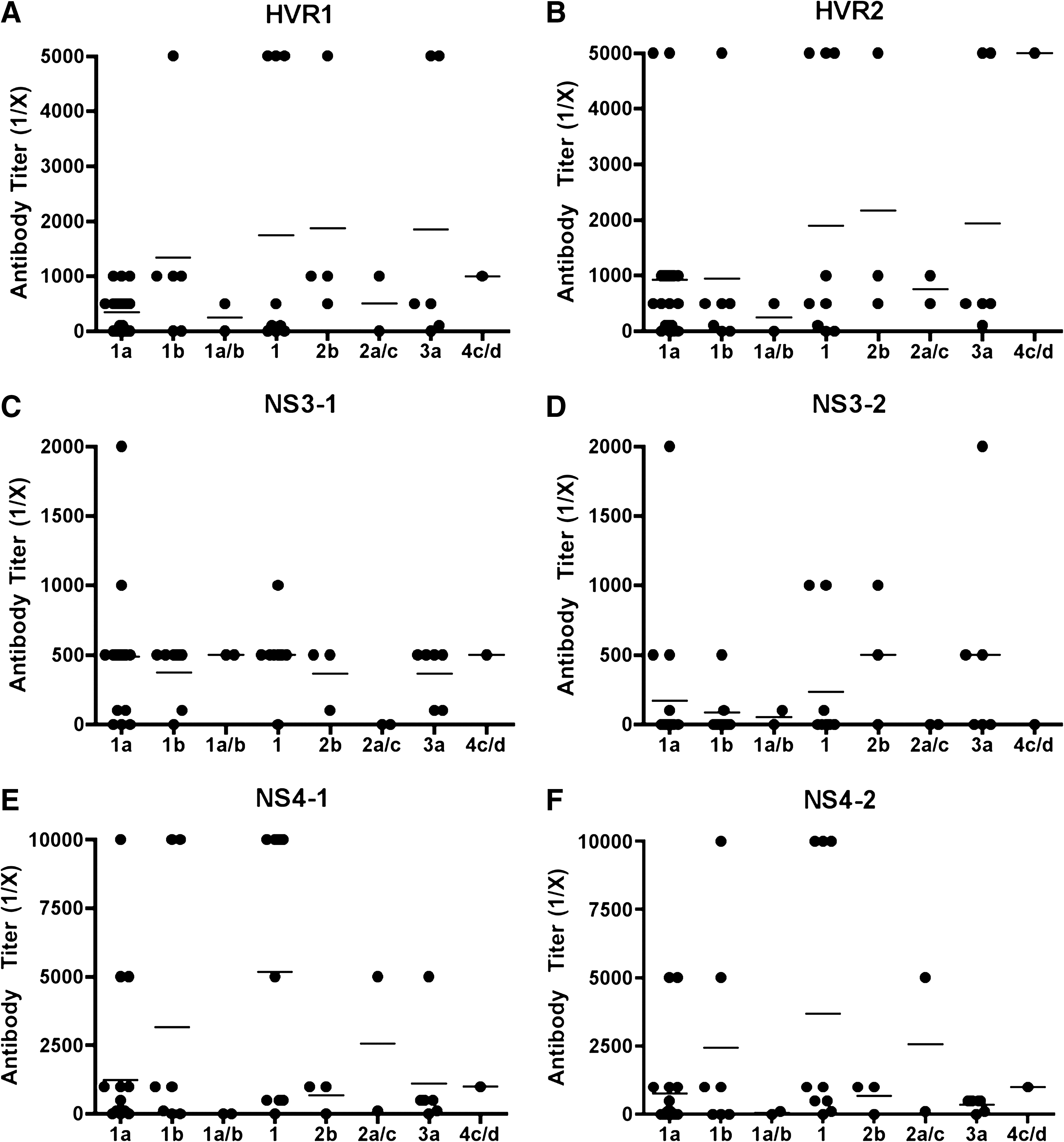
Relative antibody titers to six HCV HECs in 50 plasma samples from HCV-positive patients (group 2). Recognition of HVR1, HVR2, NS3-1, NS3-2, NS4-1 and NS4-2 HCV HECs by patients infected with different genotypes of HCV (1a, 1b, 1a/b, 2b, 2a/c, 3a, and 4c/d). Each dot represents an individual plasma sample from an HCV patient. Antibody end-point titers for binding to HCV HEC were determined based on the OD value of normal human sera used as negative control.
Includes subtypes 1a, 1b, 1a/b, and genotype 1 of unknown subtype.
Includes subtypes 2b and 2a/c.
Includes subtype 3a only.
Abbreviations: HEC, hypervariable epitope construct; HCV, hepatitis C virus; HVR, hypervariable region.
There were no significant differences in the recognition of epitopes from envelope proteins (HVR1 and HVR2) and non-structural proteins (NS3-1, NS4-1, and NS4-2), except NS3-2. More than 72% of HCV-positive patients had antibody responses to one of the HCV HECs, and 36% to NS3-2, as shown in Table 4. Patients infected with genotype 1 showed lower antibody titers to NS3-2 HEC than to the other HECs. This outcome could be due to the weak antibody response to the natural epitope represented by NS3-2 HEC. These results suggest that the NS3-2 HEC epitope may induce a cellular rather than a humoral response against HCV. Compared to the antibody binding to the NS3-2 HEC, the NS3-1 HEC was the most recognizable epitope (86%) among the HCV patient plasma samples tested. However, 80% of patients (34/43) had low antibody titers (1:500). This implies that the NS3-1 HEC epitope might induce production of constant but low levels of antibodies, in addition to cellular responses against HCV.
The NS4, NS4-1, and NS4-2 HECs represent 32 sequence variants of the epitope. Some patients had very strong antibody responses (1:10,000 titer) directed at NS4-1 (14%) or NS4-2 (8%). Interestingly, the genotype of these HCV-infected patients was 1 (1a, 1b, or unknown subtype of 1), and the viremia was more than 500,000 IU/mL. In addition, NS4-1 HEC was preferentially recognized over NS4-2 HEC by patients with chronic HCV infection (Fig. 2).
The antigen-specific humoral response to HCV HECs
Immunogenicity of HEC constructs was also studied in mice. Individual HEC-specific antibody responses from mice immunized with a mixture of six HECs (HVR1, HVR2, NS3-1, NS3-2, NS4-1, and NS4-2) were analyzed. Sera obtained before immunization were used as negative controls.
The antibody response to HVR2 HEC was very strong (1:100,000 titer), up to 100 times more than the antibody titer to HVR1 HEC. The antibody responses to HECs derived from non-structural proteins NS3 and NS4 were also stronger (1:4,000 antibody titer) than to HVR1, but not as high as that of HVR2 HEC. These results suggest that all of the HCV HECs induce strong antibody responses in mice, and that HVR2 HEC represents an immunodominant epitope in terms of antibody response (Fig. 3A).
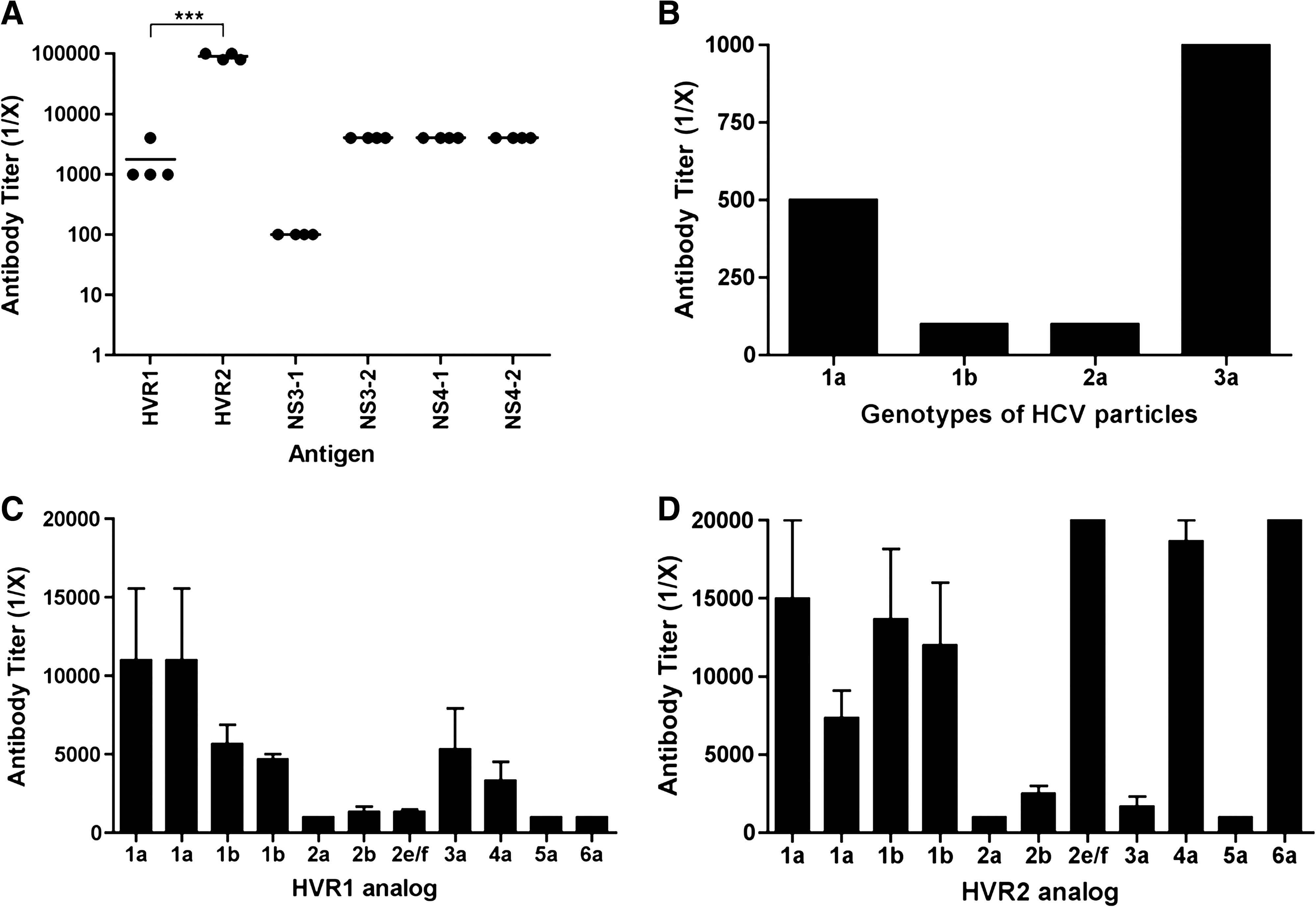
Immunogenicity of HCV HECs in BALB/c mice. (
Next, we assessed if antibodies from mice immunized with HCV HEC were able to recognize HCV circulating in patients with HCV chronic infection. The use of recombinant E2 protein and synthetic HCV virus-like particles was replaced with virus particles purified from plasma of patients infected with HCV. In order to test mouse antibody binding activity to viral particles derived from HCV patients with different genotypes (1a, 1b, 2a, and 3a), 10 μg of particles per well were plated as coating antigen on ELISA plates. Mouse serum samples obtained before immunization were used as negative controls to determine the titer. Interestingly, viral particles from all genotypes tested were recognized by sera from mice immunized with a mixture of the six HCV HECs, up to 1:1000 dilution in the case of genotype 3a viral particles (Fig. 3B).
Cross-reactivity of antibodies from mice immunized with a mixture of six HCV HECs was also assessed (Fig. 3C and D). Pre-bleed serum samples were used as negative controls to determine antibody titer. Mouse serum samples were tested and diluted until the antibody titer was up to 1:20,000 in cases requiring further dilution. We concluded that sera from mice immunized with a mixture of HCV HECs had cross-reactive antibodies that could recognize all the HVR1 and HVR2 analogs (antibody titers to 1:1000). In addition, cross-reactivity of antibodies against HVR2 analogs was superior to that against HVR1 analogs, except for three of the analogs tested (2a, 3a, and 5a).
Lymphocyte proliferation
HCV HEC-specific lymphocyte proliferation was determined by thymidine incorporation. Six outbred mice (CFW) per group were immunized with two HECs combined, or a mixture of all six HECs. The two-HEC combinations were (1) HVR1 and HVR2, (2) NS3-1 and NS3-2, and (3) NS4-1 and NS4-2. An unrelated peptide (SIV) was used as a negative control. In the group immunized with the HVR1 and HVR2 HECs (Fig. 4A), the proliferation of HVR1-specific lymphocytes was approximately three times stronger than that of HVR2 HEC and SIV HEC. Antibody-binding studies in mice showed that the epitope represented by HVR2 HEC induced a stronger humoral response than that represented by HVR1 HEC (Fig. 3A). These two sets of results correlate, suggesting that the weak humoral response is not due to the absence of antigen-specific antibodies, but is because the immune response is skewed toward a cellular response. It could also suggest that the HCV HECs are strong immunogens capable of inducing cellular as well as humoral responses.
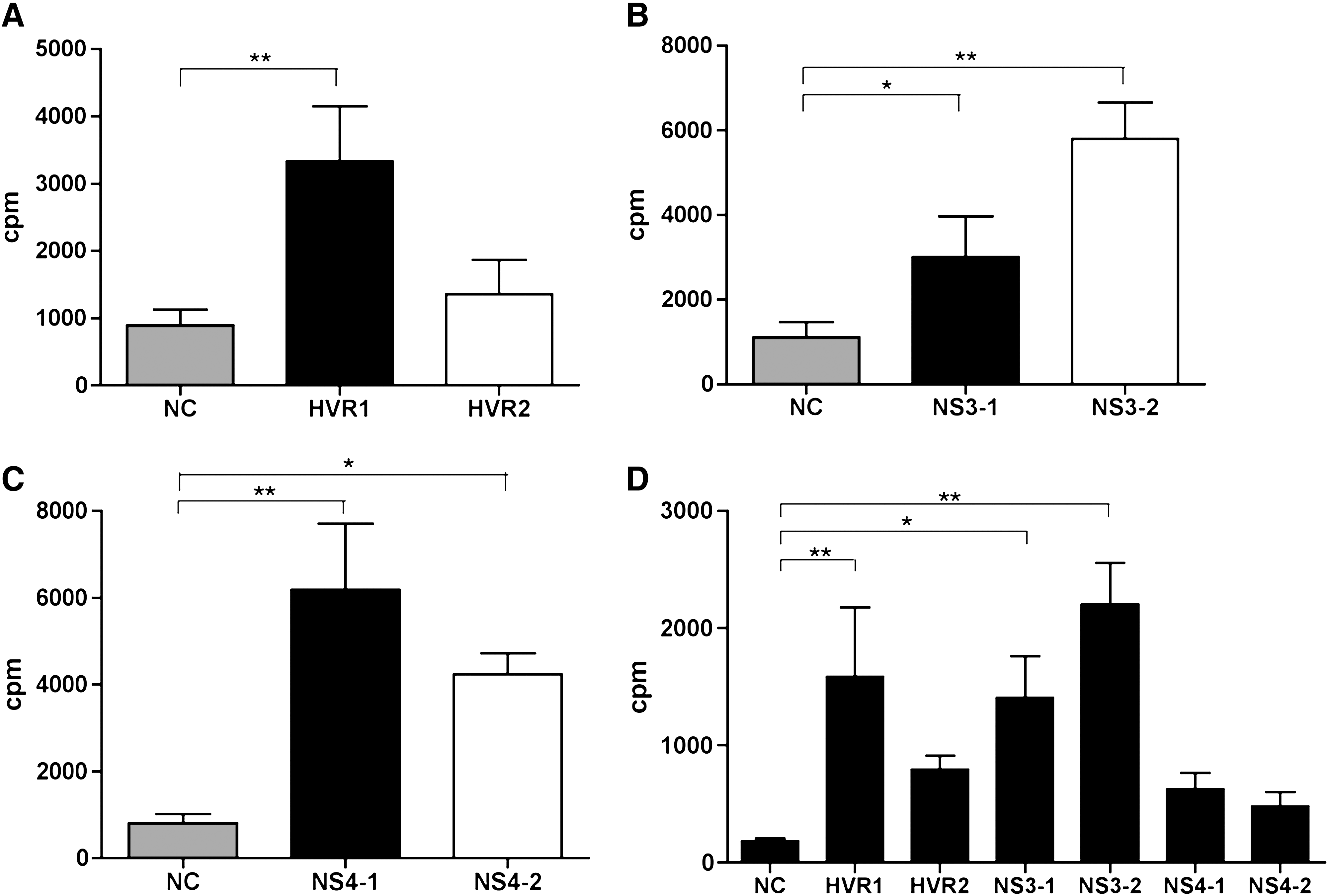
HCV HEC-specific lymphocyte proliferation as measured by thymidine incorporation. Six outbred mice (CFW) per group were immunized with a combination of two HECs, or with all six HECs together. The two-HEC combinations were (1) HVR1 and HVR2, (2) NS3-1 and NS3-2, and (3) NS4-1 and NS4-2. An unrelated peptide derived from SIV was used as a negative control (NC). Lymphocytes from lymph nodes were stimulated in vitro with each HCV HEC at 5 μg/mL for 3 d. Following 14 h of incubation with 1 μCi of tritiated thymidine per well, the incorporated thymidine was determined and is presented as counts per minute (cpm). (
Although both NS3-1 and NS3-2 HECs induced antigen-specific lymphocyte proliferative responses, the NS3-2 HEC was a more dominant epitope than the NS3-1 HEC (Fig. 4B). Both NS4-1 and NS4-2 HECs induced strong cellular responses (Fig. 4C). The level of proliferation (in cpm) was at least four times higher than that of the SIV negative control (Fig. 4C).
HEC-specific lymphocyte proliferation was also tested in the group immunized with a mixture of all six HECs (Fig. 4D). A response to each individual HEC was detected (at least twice than the negative control). The same pattern of proliferation responses was observed as in the group immunized with HEC pairs. The response to HVR1 HEC was stronger than that to HVR2 HEC, that to NS3-2 HEC was stronger than that to NS3-1 HEC, and that to NS4-1 HEC was stronger than that to NS4-2 HEC. However, proliferation against non-structural protein 4 in the group immunized with a mixture of HECs was not as strong as in the group immunized only with the NS4-1 and NS4-2 HECs.
Detection of cytokine production
We assessed IFN-γ, IL-2, and IL-4 cytokine secretion from HEC-specific T lymphocytes by cytokine ELISA. We observed similar results to those obtained from HEC-specific proliferation responses. IFN-γ and IL-2 were detected in response to HVR1, NS3-2, NS4-1, and NS4-2 HECs in the groups immunized with the two-HEC combinations. The amount of IFN-γ and IL-2 detected in the HVR2, the NS3-1, and the SIV negative control was either undetectable or less than 35 pg/mL. IL-4 was not detected in any group except for HVR1 HEC (14.9 pg/mL) in the group immunized with the two-HEC combinations (Fig. 5A, B, and C). In the mice immunized with a mixture of all HECs, IFN-γ and IL-2 were detected in individual HECs, except for the HVR2 HEC, in which the amount of IFN-γ was the same as the negative control (about 25 pg/mL) (Fig. 5D). These results suggest that HEC-specific immune responses were Th-1-mediated (cellular), and not Th-2-mediated (humoral) responses, because IL-4 secretion was either minimal or undetectable.
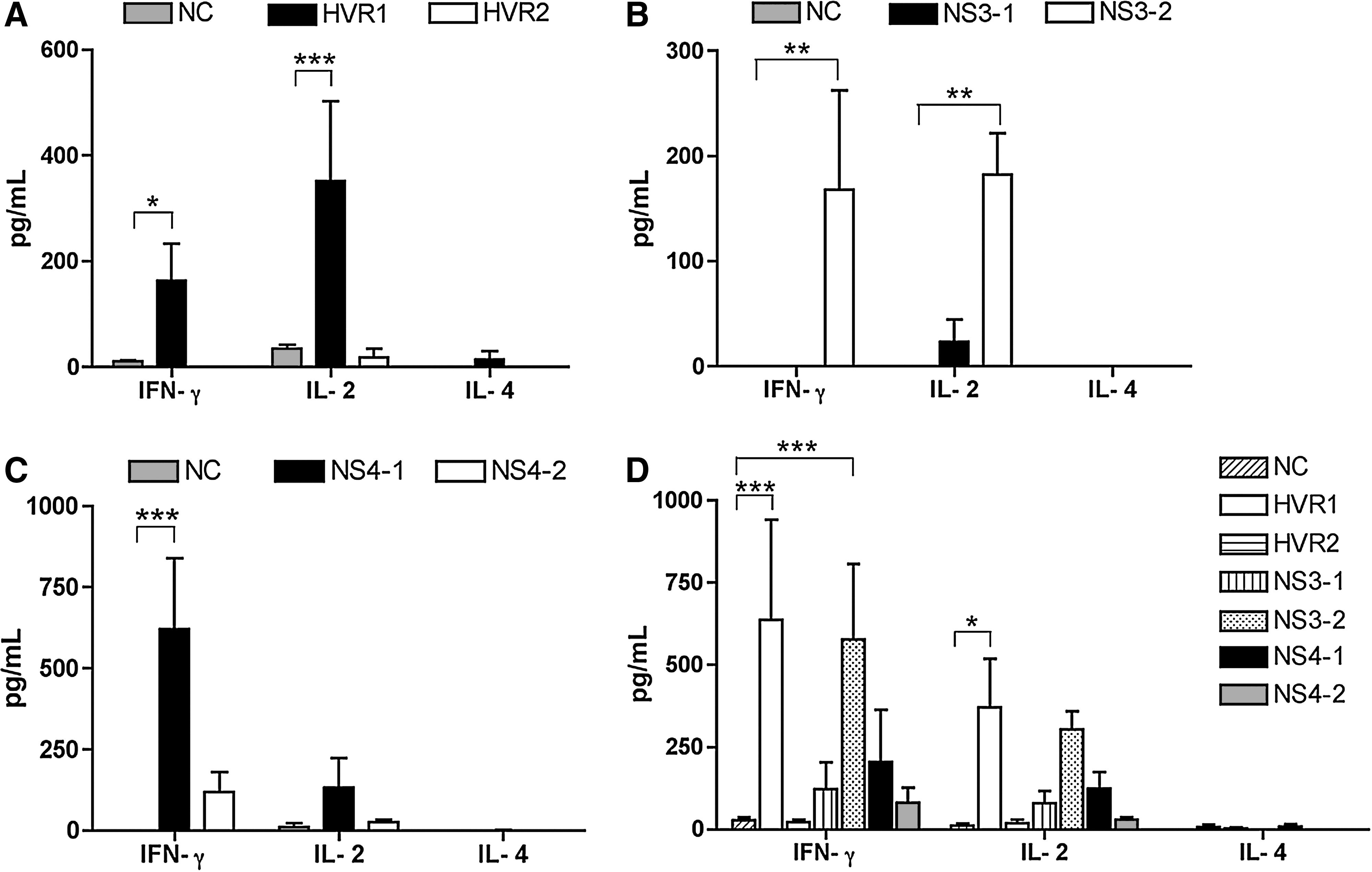
Detection of cytokines secreted from HCV HEC-specific lymphocytes. IFN-γ, IL-2, and IL-4 cytokines secreted from HEC-specific T lymphocytes were quantitated by cytokine ELISA. In each group, 5 × 105 lymphocytes from the spleens were stimulated in vitro with each HCV HEC at 5 μg/mL for 2 d, and the supernatants were collected to measure cytokine levels. An HCV-unrelated peptide (SIV) was used as a negative control (NC). (
Discussion
Numerous studies have contributed over several decades to our understanding of cellular immune responses during both chronic persistent and self-resolved HCV infections (5,16,30,40). Although a protective CD8+ CTL response plays an important role in the clearance of HCV infection, it is strongly suggested that a robust CD4+ T-helper response is also essential in control of viral replication and for the promotion of effective antiviral CD8+ CTL responses (1,19,33). In addition, HCV-specific cellular responses during antiviral treatment in patients with HCV chronic infection have been extensively studied (7,13). In contrast to the detailed understanding of cellular immune responses upon HCV infection, humoral immune responses for the control of HCV infection have not been well analyzed. Humoral immune responses produce antibodies that are broadly cross-reactive to antigenic variants of epitopes. Chronic HCV infection is usually established regardless of the antibody production that targets various epitopes of the HCV structural and non-structural proteins. Neutralizing antibodies play an important role in controlling hepatitis C viral infection and dissemination by directly blocking attachment and entry of HCV to host target cells (25,35,41). A recent study showed that human monoclonal antibodies were broadly reactive to neutralize heterologous HCV isolates. This study also demonstrated protection against HCV quasispecies infection by neutralizing antibodies in a human liver chimeric mouse model (20).
To what extent antibodies from an individual HCV patient are broadly reactive at the epitope level among different genotypes and subtypes, and their specificity for epitopes derived from different regions of the viral proteins remains unclear. Also the clinical significance of these anti-HCV antibody responses remains unknown. Our study showed that high antibody titers to HECs representing variable antigenic epitopes of both envelope and non-structural proteins were observed in all HCV patients. This implies that patients with HCV chronic infection develop broadly cross-reactive antibodies against all viral proteins, but these antibodies are not able to control the infection. These data may suggest that significant amounts of tissue destruction and viral lysis occur in vivo, and that the structural viral proteins that are released could drive these antibody responses. Based on these results we are unable to determine to what extent antibody responses are protective and driving immune pressure against intact virions, or are simply a consequence of tissue destruction due to immune pressure mediated by CTLs, or both. However, the data indicating that the majority of patients with chronic HCV infection had antibodies to the hypervariable epitopes of HCV envelope protein suggest that sequence mutation of HVR1 epitopes occurs as a result of immune pressure–induced cross-reactive antibodies against different variants of the HVR1 epitope. This resulted in higher antibody titers to the corresponding sequences represented within the HVR1 HEC designed for our study.
The NS3 protein has two enzymatic functions: serine protease at the amino-terminal (180 amino acids) (31), and NTPase/RNA helicase at the carboxy-terminal (450 amino acids) (18). Although the HCV genome is highly variable, core and NS3 proteins undergo less antigenic variability than the two envelope glycoproteins (E1 and E2) and other non-structural proteins (27). A report suggested that NS3 interrupts TLR3 pathways by degradation of toll/IL-1 receptor domain-containing adapter inducing IFN-β (TRIF) using NS4A as a serine protease cofactor facilitating HCV evasion of the immune response (21). Therefore, lysis of cells expressing NS3-specific epitopes could be involved in viral clearance by the host immune response. A strong cellular immune response induced against NS3 is considered to play an important role in viral clearance. A previous study found that the strongest and most consistently detected CD4+ T-helper cell response was to NS3 protein in patients with self-resolved HCV infection (14). This study also suggested that NS3 protein contains identified dominant epitopes for CD4+ T-helper responses in both humans and chimpanzees with resolved infection (30,39). In addition, one of the CD4+ T-cell epitopes recognized by more than 30% of HCV patients with spontaneously resolved infection as determined by cell proliferation assays and IFN-γ detection was also from the same region as our HCV NS3-2 HEC (15,29). It was interesting to detect low titers of antibodies that bind NS3-1 and NS3-2 HECs in patients with chronic HCV infection. More interestingly, our NS3-1 and NS3-2 HECs contained similar sequences of identified CD4+ T-cell epitopes from NS3 to those used by other groups (15,30,39). We concluded that the reason for low antibody titers to NS3-1 and NS3-2 HECs was because these HECs could be epitopes for CD4+ T-cell responses. Another possibility could be that weak or absent CD4+ T-cell responses against NS3 protein in patients with chronic HCV infection affect production of large amounts of NS3-specific antibodies, and thus have low affinity antibodies to NS3 epitopes.
We designed NS4-1 and NS4-2 HECs to represent two different regions of the NS4 proteins. NS4-1 and NS4-2 HECs are composed of the C-terminal part of the NS4a protein (aa 1686–1711), and the N-terminal part of the NS4b protein (aa 1711–1733), respectively. Although we designed our own antigenic variable epitopes from NS4 using Genbank sequences, we found that other laboratories designated those two specific regions of the NS4 protein as region 5-1-1 (10). When sera from patients in group 2 were tested for binding to all six HCV HECs, we found that the antibodies were more reactive with NS4-1 HEC than NS4-2 HEC. According to a previous study, the peptide variants of NS4a region 5-1-1 were broadly recognized by patients infected with different genotypes, whereas the peptide variants of NS4b were only bound by antibodies from patients infected with the same genotype (10). Interestingly, these peptide variants contained the sequence PDREVLYQEFDE, that is also part of our NS4-1 HEC. This may explain why antibodies to NS4-1 HEC were more frequently detected than to NS4-2 HEC in plasma from HCV-infected patients. Another group also noted that antibodies to NS4 were more frequently found in patients infected with genotype 1 than other genotypes (12). Epitopes derived from NS4 were considered among the most immunogenic for induction of cellular responses against HCV, when tested for induction of proliferation of human cells from patients with chronic HCV infection (28). This might explain why patient antibody binding to NS4 was higher than to the other epitope variants tested in the present study.
We showed that HCV HECs are strong immunogens that induce both humoral and cellular immune responses in mice. The antibody response induced by HCV HEC in mice suggests that HVR2 HEC is an immunodominant epitope that drives a humoral response. In addition, HVR2 HEC-specific antibodies produced in mice immunized with a mixture of six HCV HECs were broader and stronger than the response to the HVR1 analog. Lymphocyte proliferation measured with the thymidine incorporation assay and cytokine ELISA to assess levels of IFN-γ, IL-2, and IL-4 support our conclusion. In addition, HEC-specific cellular responses were also detected in CFW outbred mice that represent individual MHC polymorphisms. Specific lymphocyte proliferation to HVR1 HECs was three times higher than negative controls, and twice that to HVR2 HECs. HVR1 HEC-specific IFN-γ and IL-2, but not HVR2 HEC-specific cytokine production, was observed. The results indicate that HVR1 is a more immunogenic epitope in mice than HVR2 for induction of cellular immune responses as represented by the E2 envelope glycoprotein-derived HCV HECs. In addition, the frequency of CD69+ CD4+ activated T cells in mice immunized with a mixture of HCV HECs showed that HVR1 HECs induced more activated T cells than HVR2 (data not shown). In terms of non-structural protein-derived HCV HECs, NS3-2 and NS4-1 were more immunogenic epitopes than NS3-1 and NS4-2 as measured by cellular responses. In the study designed to determine HEC-specific cytokine production, IFN-γ and IL-2 production were detected, but not IL-4, suggesting that HCV HEC-specific T-cell responses were biased to a Th-1 rather than to a Th-2 response.
Due to the unavailability of lymphocytes from HCV-infected patients, we were not able to study cellular responses to these epitopes after incubation with HCV HECs in vitro. At this point, we do not know if the epitope represented by HVR1 HECs might also induce stronger cellular responses than those represented by HVR2 in HCV patients, as observed in the mouse studies. However, there could be a correlation between our studies with human samples and immunogenicity of HCV HECs in mice. Our results showed that antibodies from HCV patients showed less recognition of NS3-2 HECs than other HCV HECs. Previous studies that have reported T-cell responses induced by epitopes derived from the NS3 region (15,30,39) correlate with our mouse data showing that NS3-2 HECs induce less immunogen-specific lymphocyte proliferation and Th-1-biased cytokine production than other HCV HECs.
Overcoming antigenic variation with a vaccine representing only a single genotype of HCV will probably be impossible. However, HECs can represent many variant forms of viral isolates rather than having to synthesize a large number of peptides representing each divergent form of each antigenic epitope. Like other peptide-based vaccines, reversion to virulence, cold preservation requirements, and other negative implications associated with live-attenuated virus vectors are not a concern. In addition, HECs designed at our laboratory are strong synthetic peptide immunogens inducing both humoral and cellular immune responses directed to the most variable HCV epitopes. We were not able to perform an HCV challenge efficacy study with animals immunized with HCV HECs due to the lack of susceptible small animal models for HCV. Neutralization assays were not performed due to the lack of appropriate detection antibodies. However, all of the human and mouse data obtained suggest that HECs represent antigenic epitopes being recognized by more than 80% of HCV patients infected with diverse genotypes, and could have potential as peptide components of vaccine candidates, as they induce both humoral and cellular immune responses specific to HCV. Therefore, HECs could be used to develop potential peptide vaccine components to overcome antigenic variation against viruses with mutating genomes such as HCV.
Author Disclosure Statement
The authors have no conflicts with regard to financial interests. This material has not been previously reported and is not under consideration for publication elsewhere.