
Abstract
The purpose of this study was to screen and identify the linear B-cell epitopes of Epstein-Barr virus (EBV) latent membrane protein 2 (LMP2). The secondary structure and surface properties of EBV LMP2A protein were analyzed. In combination with hydrophilicity, accessibility, flexibility, and antigenicity analysis, and average antigenicity index (AI) of epitope peptide investigation, three peptides were selected as potential candidates of linear B-cell epitopes. The peptides were 199–209 (RIEDPPFNSLL), 318–322 (TLNLT), and 381–391 (KSLSSTEFIPN). The fragments encoding potential B-cell epitopes were cloned and overexpressed in an E. coli system. The immune sera of these fusion proteins were collected from BALB/c mice by subcutaneously immunizing them three times. Western blotting results showed that these epitope recombinant proteins could be recognized by the serum antibodies against the whole LMP2 from nasopharyngeal carcinoma (NPC). Indirect ELISA measuring individual sera from 196 NPC patients, 44 infectious mononucleosis (IM) patients, 253 healthy adults, and 61 healthy children, indicated that NPC patients had significantly higher reactivity to these epitope-fused proteins compared with IM and healthy individuals (p < 0.05). In addition, all the immune sera of peptide-fused proteins responded to native LMP2A antigen obtained from the EBV prototype strain, B95-8 cells. IFA results confirmed that specific antibodies induced by epitope peptide-fused proteins recognized intracellular regions of LMP2A. These results demonstrated that these three predictive epitopes not only were immunodominant B-cell epitopes of LMP2A, but also may be potential targets for applications in the design of diagnostic tools.
Introduction
Accumulating evidence implies that EBV oncogenesis is principally associated with latency status. During latency status of EBV infection, only a limited subset of the full repertoire of viral genes is transcribed, including six nuclear proteins (EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA-LP), and three membrane proteins (LMP1, LMP2A, and LMP2B) (5). LMP2A and LMP2B are two alternative forms of the hydrophobic membrane protein LMP2(23), which are transcribed across the fused terminal repeats of the EBV episome by alternative promoter usage. Their mRNAs include eight common exons and one 5′ exon unique to each type. The first exon of LMP2B is noncoding, whereas that of LMP2A encodes an additional N-terminal 119-aa cytoplasmic domain (2). Their identical structural properties include 12 hydrophobic membrane-spanning domains and a 27-amino-acid cytosolic carboxy terminus. The consistent detection of LMP2A and LMP2B in EBV-associated malignancies such as HD, NPC, and gastric adenocarcinoma suggests that these viral proteins may play a role in disease pathogenesis. It has been demonstrated that LMP2A may cooperate with the aberrant host genome, and thereby contribute to malignant transformation by intervening in signaling pathways, especially the cell cycle and apoptotic pathway (26). Moreover, LMP2B co-localizes with LMP2A (24), and both of them can promote epithelial cell spread and motility (1).
Detailed analysis of LMP2 epitopes is important for both the understanding of immunological events and the development of epitope-based vaccines, as well as diagnostic and therapeutic tools for EBV infections. Recently, several HLA-restricted CTL epitopes in LMP2 protein have been identified (17,28,30). Moreover, currently available monoclonal antibodies target epitopes in the intracellular regions of LMP2, and might not bind the surface of EBV-infected cells (6), so they are not suitable for antibody-mediated diagnosis and therapy. Therefore, the aim of the present study was to screen and identify the novel B-cell epitopes in the extracellular regions of EBV LMP2A protein, using a bioinformatics method in combination with molecular biology methods.
Materials and Methods
Epitope prediction
The complete amino acid sequence of EBV LMP2A was retrieved from the SwissProt Database (
Expression and purification of epitope peptide-fused proteins
Expression and verification of recombinant proteins
To express the predicted peptides, the amino acid sequence of each peptide was translated into DNA sequences using codon usage of a prokaryotic system. The sense and antisense strands targeting each peptide were synthesized and designed to include BamH I and Xho I restriction sites at the 5′ and 3′ end, respectively. After sense and antisense strand oligos were annealed and confirmed, the fragments were cloned into the BamH I and Xho I sites of pET32a(+). A 6 × His tag was added to the N and C terminus of these fragments to facilitate purification. After confirmation of the inserted sequences by enzyme digestion and DNA sequencing, positive plasmids were transformed into E. coli BL21 (DE3) for expression. After 1 h of shaking, isopropyld-thiogalactopyranoside (IPTG; Sigma-Aldrich Co., St. Louis, MO) with a final concentration of 1 mM was added into Luria-broth (LB) medium with ampicillin (100 mg/L), and a further 3 h of agitation was introduced. Then the bacteria cells were pelleted at 10,000 g for 15 min and lysed by sonication in phosphate-buffered saline (PBS; pH 7.4). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out to analyze cell lysates containing 6 × His-tagged recombinant proteins with Coomassie blue staining. To verify the presence and apparent molecular size of the recombinant proteins, Western blotting was carried out using anti-His antibody (Sigma-Aldrich), and detected with 0.005% (w/v) 4-chloro-1-naphthol and 0.015% (v/v) hydrogen peroxidase.
Purification of recombinant proteins
E. coli-synthesized fused protein with 6 × His-tag was produced as inclusion bodies in the cell lysate. For purifying proteins expressed in E. coli as insoluble particles, a pellet obtained from a 50-mL culture was resuspended in 5 mL of buffer B (100 mM NaH2PO4 and 10 mM Tris · Cl; pH 8.0) containing 8 M urea and 20 mM PMSF and lysed by ultrasonication on ice. After centrifugation at 12,000 g for 10 min, the recombinant protein was purified by affinity chromatography using a precharged Ni-NTA Sepharose column (Qiagen Inc., Valencia, CA). The recombinant protein was then eluted with a linear gradient imidazole (from 5 to 500 mM). Fractions were collected and analyzed by 12% SDS-PAGE. Fractions containing the recombinant protein were pooled and dialysed to remove imidazole and urea. Dialysis was performed in PBS-T buffer (0.05% Tween 20; pH 7.4) that contained decreasing concentrations of urea (6–0 M). Dialysis purity was verified by SDS-PAGE, and protein concentration was determined by the bicinchoninic acid (BCA) protein quantitation method. Samples were stored at −20°C until use.
Preparation of immune sera of epitope peptide-fused proteins
Antisera to the epitope peptides were produced by subcutaneously inoculating BALB/c mice three times with 100 μg purified His-tagged recombinant protein at 3-wk intervals. All antigens were emulsified with an equal volume of Freund's incomplete adjuvant just before injection, except for the first dose with Freund's complete adjuvant. Sera were collected prior to the first immunization and 2 wk after each immunization, and all the sera were analyzed with serologic assays or stored at −20°C until testing.
Preparation of native antigen of LMP2A
Native LMP2A protein was obtained from EBV B95-8 cells (ATCC CRL-1612) and cultured in RPMI 1640 containing 10% fetal bovine serum (FBS). Then 1 × 107 cells in the exponential phase of growth were spun and washed several times with PBS (pH 7.0). Then cycles of freezing and thawing of the cells was repeated, following ultrasonication for 5 min on ice. To reduce protein degradation, 20 mM PMSF was added during this process. After centrifugation at 12,000 g for 20 min, the protein solution in the upper aqueous layer was carefully collected and stored at −20°C until further analysis.
Collection of sera
Patient characteristics are shown in Table 1. In all, 196 NPC patients (151 men and 45 women) 24–75 y old (median age 41 y) were selected from the Department of Otorhinolaryngology, the First Affiliated Hospital of Wenzhou Medical College (Zhejiang Province, China) between January 2009 and January 2010. All NPC patients were newly diagnosed and pathologically confirmed. The stage of disease progression was classified according to the 1996 Union International Cancer Control classification. The NPC group included 12 cases at cancer stage I, 32 at stage II, 98 at stage III, and 54 at stage IV. The sera of 253 healthy adults, matched by age and sex, were selected after physical examination. Forty-four IM patients, identified on clinical grounds and by heterophile Ab positivity, were selected from the Department of Hematology, Yuying Children's Hospital of Wenzhou Medical College. In all 61 healthy children, matched by age and sex, were chosen after physical examination. Informed written consent was obtained from each patient and the study was approved by the Human Research Ethics Committee of the First Affiliated Hospital of Wenzhou Medical College. All sera from blood donors were collected and stored at −20°C until further analysis.
Data are sample volumes in this study.
Western blotting analysis
To evaluate the antigenicity of the expressed epitope-fused proteins, the purified samples were subjected to 12% SDS-PAGE and Western blotting analysis. 6 × His protein purified from E. coli BL21 transformed with pET-32a(+) blank vector was used as control. For immunoblot reaction, the proteins were electrophoretically transferred from gels onto nitrocellulose membranes. After blocking with 5% (w/v) nonfat milk powder in TBS, the membranes were reacted with a 1:100 dilution of EBV-positive NPC sera in 5% (w/v) nonfat milk powder in TBS. The membranes were further incubated with HRP-conjugated goat anti-human immunoglobulin G (IgG), and color reaction was developed with 0.005% (w/v) 4-chloro-1-naphthol and 0.015% (v/v) hydrogen peroxidase color development substrate.
Indirect ELISA analysis
To evaluate the ability of immune sera of epitope-fused proteins to recognize the LMP2A antigen, microplates were coated with 10 μg/mL of B95-8 cell lysate (about 1 × 105 cells) in carbonate coating buffer (100 μL/well). Microplates coated with 2 μg/mL of epitope peptide-fused proteins were used to evaluate the reactivity of individual sera from NPC patients, IM patients, and healthy individuals to these epitope peptides. After incubation at 4°C overnight, the plates were rinsed with PBS solution containing 0.05% Tween 20 (PBST). The coated wells were blocked by 100 μL blocking buffer (PBST containing a 3% solution of nonfat milk powder) at 37°C for 1 h. The plates were washed 5 times with PBST after blocking, and 100 μL of serially diluted immune sera of mice and individual sera from NPC patients, IM patients, and healthy individuals was added to the wells in triplicate, and the plates were incubated at 37°C for 1 h. After washing 5 times, 100 μL of a 1:1000 dilution of the HRP-conjugated goat anti-mouse IgG or goat anti-human IgG were added to each well and incubated for 1 h at 37°C. The wells were washed again, followed by the addition of 100 μL/well of 3,3′,5,5′-tetramethylbenzidine (TMB)-H2O2 solution. The reaction was carried out at room temperature for 20 min, then stopped with 50 μL of 1 M H2SO4 per well, and the absorbance (OD) was measured at 450 nm by using a Bio-Tek ELISA microplate reader.
All samples were independently analyzed in triplicate. The data are shown with the mean ± standard error of the OD 450 absorbance value. One-way analysis of variance (ANOVA) and an independent t-test were used to evaluate the differences of the antibody levels between the different groups; p < 0.05 was considered statistically significant. All calculations were performed with SPSS 16.0 software.
Indirect immnofluoresence assay
Cultured EBV-positive B95-8 cells were collected and washed three times with 0.01 M PBS. After being fixed in 4% paraformaldehyde (PFA) for 10 min at room temperature and washed with cold 0.01 M PBST, the cells were blocked with blocking buffer (PBS containing 5% FBS) at 4°C overnight. Then the cells were incubated with 1:100 diluted immune sera of epitope-fused proteins in PBS containing 5% FBS for 2 h at room temperature. After extensive washing in cold PBST, FITC-conjugated goat anti-mouse IgG diluted 1:100 in PBS containing 5% FBS was added, and incubated for 1 h at room temperature. After extensive washing, the cells were pipetted to slides and examined under confocal microscopy.
Results
Prediction, selection, and design of B-cell epitopes
The predicted structure of LMP2 in the cell plasma membrane has been previously reported (6). The outer membrane region within LMP2 is the first factor of the potential B-cell epitope. The analysis of the transmembrane domain indicated that the regions of aa 144–150, 202–208, 265–268, 318–322, 375–390, and 445–455 in the N-terminal belonged to the outer membrane region, and the segment of 1–120 was intramembrane (6), so the predicted six outer-membrane regions were selected for further analysis. In addition, secondary structure, hydrophilicity, flexibility, surface accessibility, and antigenicity are important features of immunodominant B-cell epitopes. The secondary structure of EBV LMP2A was predicted by methods of SOPMA, GOR, nnPredict, and HNN, on the basis of the sequence of the EBV LMP2A protein obtained from the SwissProt Database (P13285). The hydrophilicity plot, flexibility plot, surface accessibility plot, and antigenicity for the EBV LMP2A protein were obtained by the methods of Hopp and Woods (11), Zimmerman et al. (31), Emini et al. (4), and Jameson and Wolf (12), respectively. These indicated that the outer membrane regions of aa 144–150, 202–208, 318–322, and 375–390 in the N-terminal contained all or most of these features, whereas the regions of aa 265–268 and 445–455 had none of these features (Table 2). Moreover, several adjacent amino acids of the outer membrane regions also participated in the predictive features of the immunodominant B-cell epitope (Table 2). So the original sequences of aa 144–150, 202–208, 318–322, and 375–390 were expanded to the regions of aa 142–150, 198–209, 317–322, and 378–391 for further evaluation of antigenic determinants using the method of Kolaskar and Tongaonkar (15). Among the regions, three peptides, aa 199–209 (RIEDPPFNSLL), 318–322 (TLNLT), and 381–391 (KSLSSTEFIPN), had high scores. The antigenic propensity value of these epitopes were 1.022, 0.837, and 1.003, respectively. Thus they were applied for further identification.
Production of 6 × His-fused peptides carrying the predicted epitopes
The DNA fragments encoding each peptide epitope were synthesized and cloned into pET32a(+). The fragment between the restriction sites of BamH I and Xho I in the pET32a(+) vector was replaced by about the same length of epitope genes. Sequencing of recombinant plasmids containing the inserts confirmed successful construction of these expression plasmids. In this study, recombinant protein expression was under the control of the T7 promoter and induced by IPTG. The cloning/expression region of the coding strand in the pET32a(+) vector contains the sequences of Trx•Tag™, His•Tag®, and S•Tag™ proteins. Thus the expected molecular weight of the recombinant proteins was about 21 kDa, which was similar to the His-tag protein produced from the empty pET32a(+) vector. The overexpression of recombinant protein results in the formation of insoluble bodies. Hence, the expressed protein was separated with 12% SDS-PAGE under denaturing conditions. Monoclonal antibody to His-Tag readily detected these bands of recombinant proteins (Fig. 1). These His-tag fusion proteins were purified by Ni-NTA agarose affinity chromatography. After the bound proteins were eluted, the purity of the final products in the portion of the eluate was approximately 95% for these recombinant proteins as determined by image analysis of Coomassie blue-stained SDS-PAGE (Fig. 2A). Using the method of BCA protein quantification, the concentration was determined to be about 5 mg/mL for all of these purified proteins.
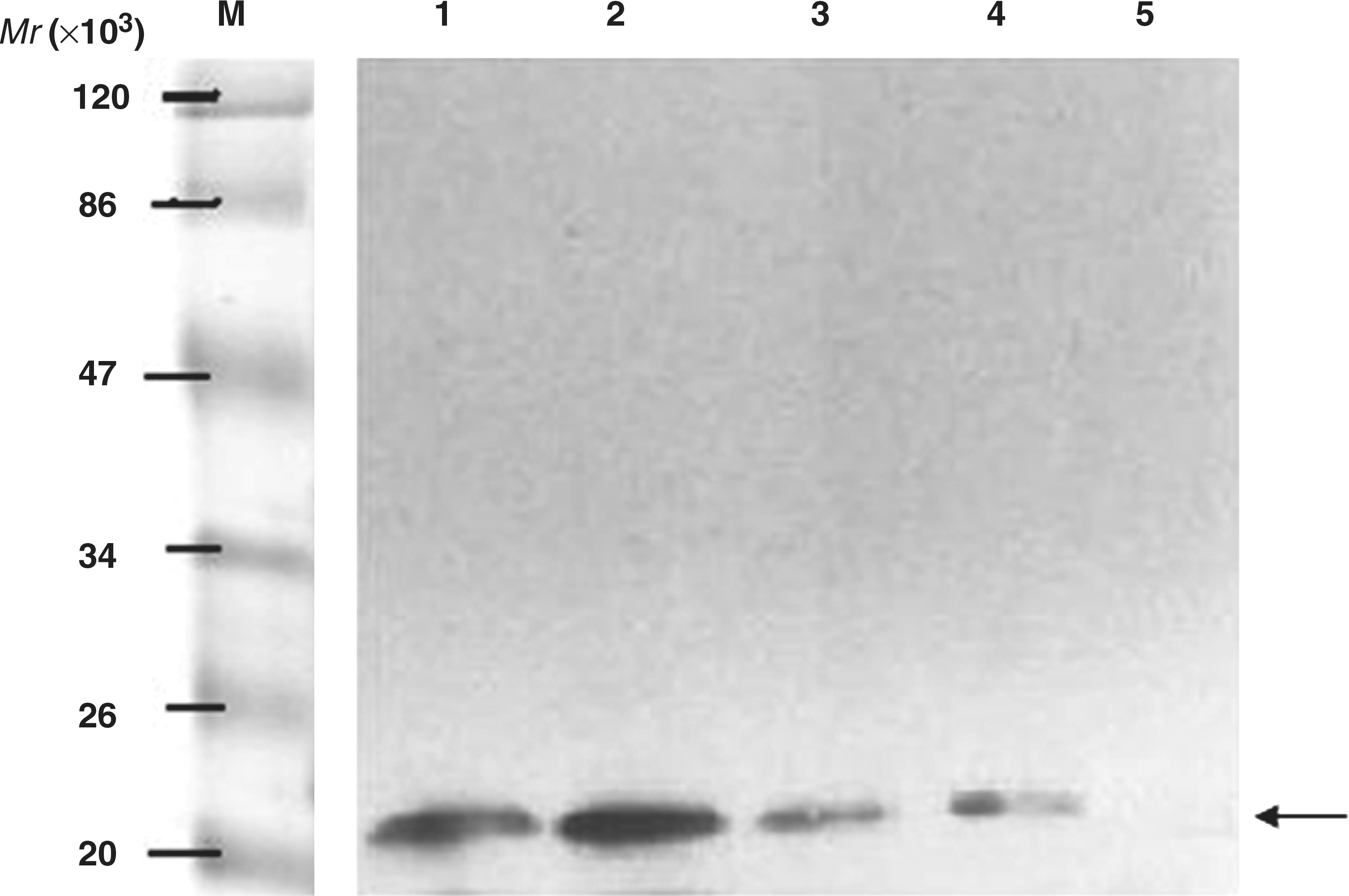
Identification of B-cell epitope fusion proteins by His-tag antibody. Lysate of induced BL21 bacteria with recombinant plasmids pET32a/epi-1, 2, and 3 and control were transferred onto nitrocellulose filters and probed with the His-tag antibody (lane 1: M, protein marker; lane 2: lysate of induced BL21 bacteria with recombinant plasmid pET32a/epi-1; lane 3: lysate of induced BL21 bacteria with recombinant plasmid pET32a/epi-2; lane 3: lysate of induced BL21 bacteria with recombinant plasmid pET32a/epi-3; lane 4: lysate of induced BL21 bacteria with empty pET32a(+) vector; lane 5: BL21 bacteria alone).
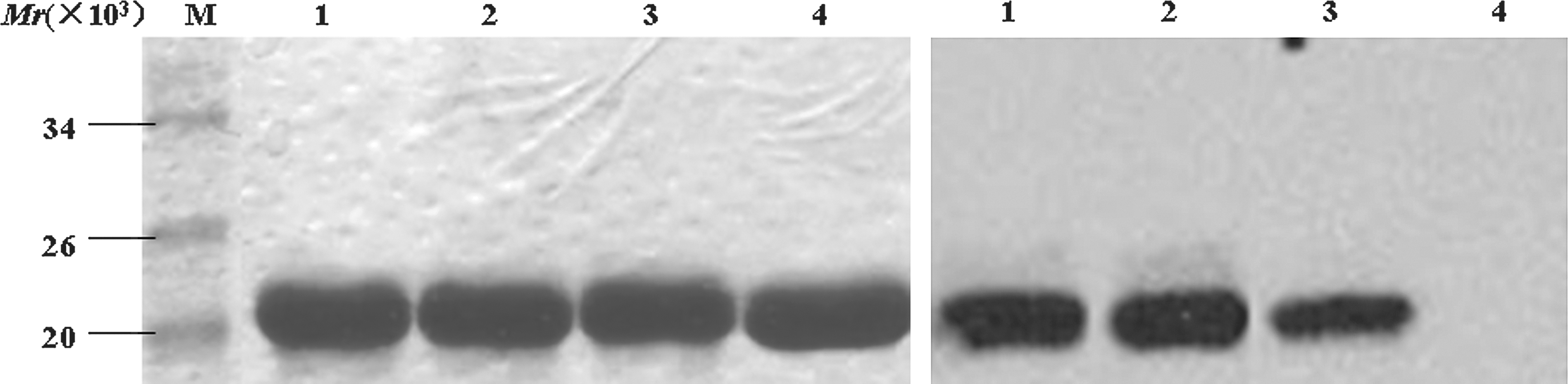
Purified recombinant proteins and epitopes identified by Western blot. (Left) The purified epitope-fused proteins and His-tag protein in 12% SDS-PAGE. (Right) Western blotting analysis of B-cell epitope fusion proteins probed with the sera mixture of 5 NPC patients (lane 1: the recombinant protein of His-tag fused epitope-1; lane 2: the recombinant protein of His-tag fused epitope-2; lane 3: the recombinant protein of His-tag fused epitope 3; lane 4: His-tag protein).
Epitope-fused proteins were recognized by sera of NPC donors
Epitopes can be identified only by their ability to bind to certain antibodies. Sera were prepared from 5 NPC donors, in which LMP2A was expressed (3,22), to test the reactivity of the epitope-fused proteins in a Western blotting assay. The purified epitope-fused proteins and His-tag were transferred onto nitrocellulose filters and probed with the sera mixture of 5 NPC patients. As shown in Fig. 2B, all 3 epitope-fused proteins could be specifically recognized by the NPC sera mixture, whereas His-tag control failed. To validate the specificity of the humoral response to these epitopes, and to obtain more convincing data on the prevalence of the epitope-specific immune responses in NPC individuals, the reactivity of individual sera from 196 NPC patients, 44 IM patients, 253 healthy adults, and 61 healthy children to these epitope peptides were further evaluated by indirect ELISA (Fig. 3). The results showed that the mean OD450 values of the recombinant proteins of His-tag fused epitopes-1, 2, and 3, in 1:800 diluted sera of NPC patients were 0.838, 0.630, and 0.502, respectively, showing significant differences when compared with IM patients and healthy individuals (p < 0.05). To evaluate the serum antibody-positive rates of epitopes in NPC, IM patients, and healthy controls, samples with higher OD450 values than the cutoff value (i.e., the mean of OD450 value of sera ± 2.1 in healthy individuals) were determined to be positive. As shown in Fig. 4, the positive rates of epitopes-1, 2, and 3 in the NPC group were 90.82%, 62.56%, and 69.39%, respectively, which were significantly higher than those in IM and healthy groups.
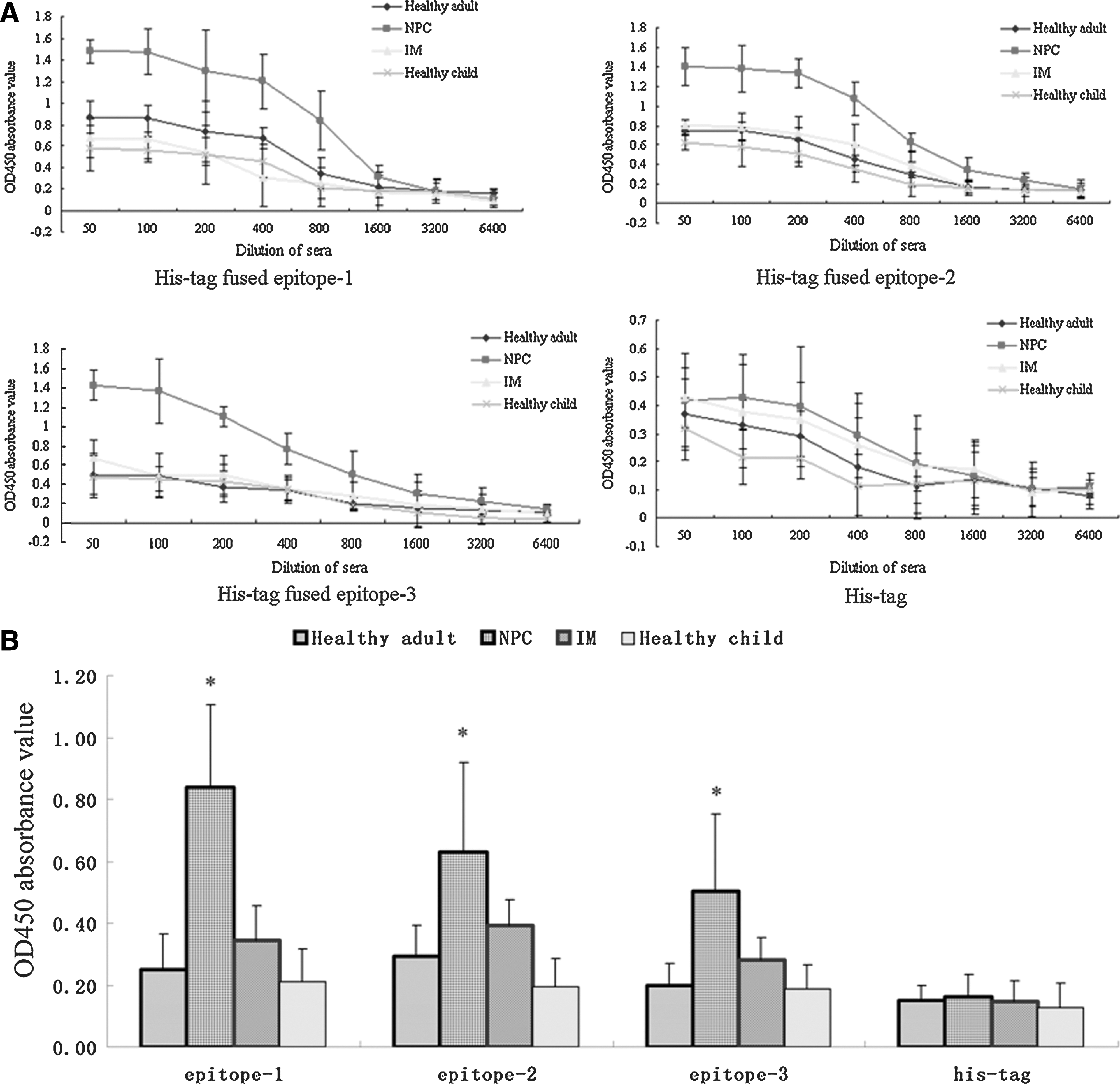
The reactivity of sera from NPC patients, IM patients, and healthy individuals to epitope-fused recombinant protein. (

Antibody-positive rates of epitopes-1, 2, and 3 in the sera of the NPC, IM, and healthy groups. The OD450 values of 1:800 diluted individual sera were greater than the cutoff value (i.e., the mean OD450 value of sera ± 2.1 standard deviations in healthy individuals) were determined to be positive.
Immune sera of peptide-fused proteins recognized native LMP2A antigen
The reactivity of immune sera of epitope-fused protein to native LMP2A antigen was further evaluated by indirect ELISA and IFA techniques. Immune sera were isolated from mice after the third immunization of epitope peptide-fused proteins. After the third immunization, specific antibodies to recombinant protein could be detected in all of these mice (data not shown). Native LMP2A antigen of EBV was obtained from EBV B95-8 cells. As shown in Fig. 5A, the OD450 of 1:50 immune sera of epitope peptide-fused proteins to EBV B95-8 cell lysate was significantly higher than the His-tag control (p < 0.05), which indicated that all induced antibodies of the fusion proteins can recognize EBV native LMP2A antigen. IFA results confirmed that extracellular regions of LMP2A were recognized by specific antibodies induced by the immune sera of epitope peptide-fused proteins (Fig. 5B). Differently from the immune sera of the B95-8 cell lysate, the fluorescence signals of the immune sera of epitope peptide-fused proteins were predominantly located on the surface of EBV B95-8 cells. These results further demonstrated that these 3 predictive epitopes were immunodominant B-cell epitopes of LMP2A.

The recognition of immune sera of epitope-fused proteins to native LMP-2A antigen. (
Discussion and Conclusion
EBV has been associated with numerous human carcinomas (27). Although the precise role of EBV in the carcinogenic process is currently poorly understood, the presence of the virus in all tumor cells provides opportunities for the development of novel therapeutic and diagnostic approaches. In case of NPC, latent membrane protein 2 (LMP2) has been found to be constitutively expressed in NPC tissue. Thus LMP2 potentially constitutes the major target antigen for diagnosis and immunotherapy of NPC. Many human leukocyte antigen (HLA) class I-restricted epitopes of LMP2 have been identified, and LMP2 is thus the most frequently recognized protein by CTLs (Fig. 6) (25). For pathogens of tumor-associated viral infectious diseases, the identification of B-cell epitopes is important for understanding the antigenic structure and interactions of virus antigen-antibody at the molecular level, and is also useful for laboratory diagnosis of associated diseases. However, based on our best knowledge there are no B-cell epitopes of EBV LMP2 reported thus far. The purpose of this study was to locate linear B-cell epitopes on EBV LMP2A, which may be potentially useful in diagnosis and immunological studies.

Schematic diagram of newly-described B-cell epitopes along with reported T-cell epitopes within the full-length LMP2A.
Epitopes can be classified into continuous (or linear) and discontinuous (or conformational) types. Methods used for mapping B-cell epitopes include region-specified PCR-mutagenesis, homolog-scanning mutagenesis, differential chemical modification of antigens, proteolysis of antigen–antibody complexes, fragmentation of proteins either by chemical cleavage or by enzymatic digestion, scans of overlapping peptides, phage display peptide libraries, x-ray crystallography studies of antibody-antigen structure, and nuclear magnetic resonance (NMR). In particular, with aid of software and databases for the prediction of epitopes, we are able to reduce the number of proteins of interest, and to decrease the number of laboratory experiments dramatically. This article described preliminary information on the novel B-cell epitopes of LMP2A obtained using bioinformatics tools and further determination by epitope mapping analysis.
Based on the use of bioinformatics, three linear epitopes on EBV LMP2A, which cover aa 199–209 (RIEDPPFNSLL), 318–322 (TLNLT), and 381–391 (KSLSSTEFIPN) in the N-terminal were highlighted. Generally, prokaryotic systems lack post-translational modifications, and do not necessarily resemble the natural proteins in a eukaryotic cell in conformation. The epitope peptides were then expressed in the prokaryotic expression system by IPTG induction. Codon usage may also vary significantly between different organisms and between genes within the same organism (13). To increase the product of the epitope-fused protein, the DNA fragments containing epitope peptides were optimized according to the codon usage of the prokaryotic system. SDS-PAGE results showed that these fused protein were efficiently expressed after being induced with 1.0 mM IPTG at 37°C for 4 h. The products of fusion proteins account for approximately 20% of the total bacterial proteins. Western blotting confirmed the fusion proteins with the monoclonal antibody of anti-His-tag. Because the fused proteins were expressed in the form of inclusion bodies, insoluble proteins accumulated in inclusion bodies were solubilized in lysis buffer containing 8 M urea, and further purified using Ni-Sepharose affinity chromatography.
We confirmed the predictive epitopes by Western blotting, IFA, and indirect ELISA. As mentioned above, sera from NPC patients contained antibodies to the entire LMP2 of EBV. If the protein of native LMP2A contained predictive B-cell epitopes, antibodies in the sera could recognize the epitope peptide-fused proteins by Western blotting. As a consequence, all of these recombinant proteins were recognized by the sera of NPC patients. Furthermore, all the immune sera of peptide-fused proteins could respond to native LMP2A antigen obtained from the EBV prototype strain, B95-8 cells, which further confirmed these 3 predictive epitopes. The data from indirect ELISA showed that all induced antibodies of the fusion proteins can recognize EBV native LMP2A antigen. Moreover, IFA confirmed that the recognition site of the specific antibodies induced by the immune sera of epitope peptide-fused proteins was in extracellular regions of LMP2A.
Indirect ELISA further showed that NPC patients had significantly higher reactivity of individual sera with these epitope peptides than IM patients and healthy individuals (p < 0.05). Positive rates of epitopes-1, 2, and 3 in the NPC group were significantly higher than those in the IM and healthy groups. These data not only further indicate that the peptides at aa 199–209 (RIEDPPFNSLL), 318–322 (TLNLT), and 381–391 (KSLSSTEFIPN) were immunodominant B-cell epitopes within EBV LMP2, but also implied that these epitopes might be potential targets for the diagnosis of NPC. The newly identified B-cell epitopes, along with the reported T-cell epitopes (9,14,16 –21) within LMP2A are shown in Fig. 6.
In conclusion, our data suggest that our methods for mapping epitopes on the LMP2A protein of EBV, which were based on bioinformatics in combination with molecular biology, are reliable and widely applicable. These results also provide a theoretical and experimental basis for the development of laboratory diagnosis of EBV infectious disease.
Footnotes
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (30972669 and 30671882), the Zhejiang Provincial Natural Science Foundation of China (Y205659), and the Wenzhou Science and Technology Bureau (Y20090248 and Y20100014).
Author Disclosure Statement
No competing financial interests exist.