
Abstract
Influenza A virus (IAV) infection is associated with outcomes ranging from subclinical infection to severe pneumonia. In this study, we compared IAV strains BJx109 (H3N2), HKx31 (H3N2), and PR8 (H1N1), for their ability to elicit innate immune responses from mouse airway cells in vitro and their virulence in mice. The viruses differed markedly in their ability to induce disease in mice (PR8 > HKx31 > BJx109). In particular, PR8 infection was associated with high levels of virus replication and pulmonary inflammation. We next compared the ability of each virus strain to infect and induce inflammatory mediators from mouse airway cells. First, major differences were observed in the ability of viruses to infect and induce chemokines and cytokines from mouse alveolar macrophages (BJx109 > HKx31 > PR8), but not from airway epithelial cells (AEC) in vitro. Second, C-type lectins of the innate immune system in mouse lung fluids blocked the ability of BJx109, but not PR8, to infect mouse macrophages and AEC. The failure of the virulent PR8 virus to elicit responses from airway macrophages, combined with resistance to antiviral proteins in mouse airway fluids, likely contribute to virulence in mice. These findings provide insight into the mechanisms underlying disease severity in the mouse model of influenza infection.
Introduction
Respiratory tract secretions contain a range of anti-microbial proteins and peptides, some of which mediate antiviral activity against IAV. In particular, surfactant protein (SP)-A and SP-D are large, oligomeric proteins of the collectin family that mediate a range of antiviral activities [reviewed in (12)]. SP-D binds in a Ca2+-depedent manner to mannose-rich glycans on the hemagglutinin (HA) and neuraminidase (NA) glycoproteins (18,20,45), whereas SP-A inhibits virus independently of calcium, acting as a sialylated γ-inhibitor that competes with cell-surface receptors for binding to the viral HA (4). The enhanced susceptibility of SP-D knockout mice to infection with highly glycosylated strains of influenza A virus highlights the important role of SP-D in innate immune defense against IAV in vivo (22,34,63). SP-A knockout mice were also more susceptible to IAV; however, the difference was not as striking as that observed in SP-D-deficient mice (33,35).
Different cell types in the lung produce differing spectrums of cytokines and chemokines following exposure to IAV. In vitro studies using human cells showed that IAV-infected epithelial cells are relatively poor producers of type 1 interferons (IFN) and proinflammatory cytokines, whereas monocytes/MΦ produce large quantities of these mediators (29,49). Furthermore, infection of lung epithelial cells favored production of IL-8 and other neutrophil-attracting chemokines (2,29), whereas human monocytes/MΦ produce inflammatory mediators that attract mononuclear leukocytes and suppress neutrophil-attracting chemokines (54). Therefore, the tropism of a particular virus strain for cells of the human respiratory tract is likely to determine the spectrum of chemoattractants produced in the lung.
Despite interest in the interactions of IAV with human epithelial cells and MΦ in vitro (2,7,9,25,29,36,49,54), few studies have addressed their murine counterparts. The mouse model of influenza infection also allows in vitro responses of airway cells to be correlated with virulence in vivo. We have demonstrated that mouse-adapted A/PR/8/34 (PR8; H1N1) infects mouse MΦ poorly and is highly virulent in mice (44,45,59). Depletion of airway MΦ has been associated with exacerbated disease (30,59,61,66); however, PR8 infection was not affected (59), indicating that a poor ability to infect airway MΦ could be an important factor contributing to the virulence of PR8 in mice. Sensitivity to innate proteins in lung fluids could further modify the ability of virus to infect and induce inflammatory mediators from airway cells. Herein, we compared PR8 (H1N1) with HKx31 (H3N2) and BJx109 (H3N2), which are reassortant viruses bearing internal components derived from PR8, in conjunction with HA and NA from A/Aichi/2/68 (H3N2) and A/Beijing/353/89 (H3N2), respectively. The three viruses were compared for (1) their ability to infect and induce inflammatory mediators from primary airway epithelial cells (AEC) and alveolar MΦ in vitro, (2) their sensitivity to neutralization by innate immune proteins in mouse airway fluids, and (3) their ability to induce cytokines and chemokines, inflammation, and disease in mice following intranasal inoculation. The virulence of the PR8 strain correlated with its ability to evade both soluble and cellular components of innate immune defense.
Materials and Methods
Mice and viruses
C57BL/6 (B6) mice and B6.RAG-1−/− mice were bred and housed in specific pathogen-free conditions at the Department of Microbiology and Immunology, University of Melbourne. Male mice 6–8 wk of age were used in all experiments. B6.RAG-1−/− mice lack mature T- and B-cell populations due to a mutation in the recombination activation gene (RAG)-1 gene (37). The IAV strains used in this study were A/PR/8/34 (PR8, H1N1), HKx31 (H3N2), and BJx109 (H3N2). HKx31 and BJx109 are high-yield reassortants of PR8 with A/Aichi/2/68 (H3N2) and A/Beijing/353/89 (H3N2), respectively, and bear the H3N2 surface glycoproteins. The viruses were grown in 10-day embryonated eggs by standard procedures and titrated on Madin-Darby canine kidney (MDCK) cells, as previously described (1).
Preparation and infection of mouse airway MΦ and epithelial cells
Mouse airway MΦ were recovered via bronchoalveolar lavage (BAL) of naïve mice, as previously described (44). Mouse AEC cultures, consisting of alveolar type I and II cells and bronchial epithelial cells, were prepared as previously described (13,15,50), with minor modifications. Briefly, lungs were excised and placed in phosphate-buffered saline (PBS) containing 100 U/mL penicillin/streptomycin. Excess tissue, including pleura and branching airways, were removed, and lungs from four mice were diced and placed in a 15-mL flask, with dissociation medium (DMEM:Ham's F-12; Invitrogen Corp., Carlsbad, CA), 1.5 mg/mL Pronase (Roche Diagnostics Corp., Indianapolis, IN), and 0.1 mg/mL DNAse I (Sigma-Aldrich, St. Louis, MO), and incubated at 37°C in 5% CO2 for 60 min. The flasks were inverted 12 times, and the contents were placed on a 70-μm filter, and cells were strained with 10 mL DMEM:Ham's F-12, supplemented with 5% fetal calf serum, 2 mM L-glutamine, 100 U/mL penicillin and streptomycin, and 120 IU/L human insulin (Novo Nordisk, Bagsværd, Denmark; DMEM/F-125). The cells were cultured in DMEM/F-125 in either 24-well plates, culture flasks, or 8-well chamber slides pre-coated with Matrigel (BD Biosciences, San Jose, CA). Cell purity was typically >99% as assessed by staining cells grown on glass cover-slips with anti-cytokeratin and anti-E-cadherin antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). LA-4 cells, a mouse respiratory epithelial cell line, was also used in some experiments, as indicated.
BAL MΦ (2.5 × 105 cells/well) or AEC (3 × 104 cells/well) were seeded into 8-well glass chamber slides, such that similar cell densities were obtained following overnight incubation at 37°C in 5% CO2. Cell monolayers were washed with serum-free medium and infected with 106 plaque-forming units (PFU) of IAV, as previously described (44). At 8 h post-infection, the slides were washed in PBS, fixed in 80% vol/vol acetone, and stained with mAb MP3.10g2.IC7 (WHO Collaborating Centre for Reference and Research on Influenza, Melbourne, Australia), which is specific for the nucleoprotein of IAV, followed by fluorescein isothiocyanate (FITC)-conjugated sheep anti-mouse immunoglobulin (Silenus, Melbourne, Australia), and viewed under 40 × magnification. The percentage of fluorescent cells was determined in a minimum of four random fields, with a minimum of 200 cells counted for each sample.
To determine the titer of infectious virus present in supernatants from IAV-infected MΦ or AEC, monolayers were incubated with 106 PFU of BJx109, HKx31, or PR8 viruses for 60 min, washed three times to remove free virus, and cultured at 37°C. At 2 h and 24 h post-infection, the supernatants were removed, and the presence of infectious virus was determined by standard plaque assay on MDCK cell monolayers in the presence of trypsin (1).
To assess the ability of BAL MΦ and AEC to produce inflammatory mediators, monolayers were infected as described above, and 24 h later the cell supernatants were collected, clarified by centrifugation, and frozen at −20°C. Levels of IFN-γ, TNF-α, IL-6, MCP-1/CCL2, and RANTES/CCL5 in cell-free supernatants were determined by mouse cytokine bead array (BD Biosciences), according to the manufacturer's instructions. Inflammatory mediator concentrations were calculated using a standard curve and expressed as picograms per milliliter. Levels of murine GM-CSF, KC/CXCL1, MIP-1α/CCL3, and MIP-2/CXCL2 were determined via ELISA (Roche Diagnostics Corp.), according to the manufacturer's instructions.
Virus neutralization assay
Neutralization of IAV infectivity was measured by fluorescent-focus reduction in monolayers of MDCK cells cultured in 96-well plates, as previously described (45).
Virus infection of mice
Mice were anesthetized and infected with 102 or 105 PFU of BJx109, HKx31, or PR8, via the intranasal (IN) route in 50 μL of PBS. The mice were weighed daily and assessed for visual signs of clinical disease, including inactivity, ruffled fur, labored breathing, and huddling behavior. Animals that had lost ≥25% of their original body weight and/or displayed evidence of pneumonia were euthanized. All research complied with the University of Melbourne's Animal Experimentation Ethics guidelines and policies. At various time points after infection, mice were euthanized and the lungs, nasal tissues, thymus, brains, and hearts were removed, homogenized in PBS, and clarified by centrifugation. Titers of infectious virus in tissue homogenates were determined by standard plaque assay on MDCK cells.
Recovery and characterization of leukocytes from mice
BAL cells were obtained as previously described (58). Samples were treated with Tris-NH4Cl (0.14 M NH4Cl in 17 mM Tris, adjusted to pH 7.2) to lyse erythrocytes, and washed in RPMI-1640 medium supplemented with 10% FCS (RF10). For flow cytometric analysis, BAL cell suspensions were incubated on ice for 20 min with supernatants from hybridoma 2.4G2 to block Fc receptors, followed by staining with appropriate combinations of FITC-, phycoerythrin (PE)-, and allophycocyanin (APC)-conjugated or biotinylated monoclonal antibodies to Gr-1 (RB6-8C5), CD45.2 (104), CD8a (53-6.7), CD4 (GK1.5), B220 (RA3-6B2), TCR-β (H57-597), and NK1.1 (PK136). Living cells were identified by the addition of 10 μg/mL propidium iodide (PI) to each sample, and the cells were analyzed using a FACSCalibur flow cytometer. A minimum of 50,000 PI− cells were collected for each sample.
Pulmonary histopathology
Hematoxylin and eosin (H&E)-stained lung sections were randomized and scored blind for airway inflammation using a scale of 0, 1, 2, 3, 4, or 5 (corresponding to none, very mild, mild, moderate, marked, and severe inflammation, respectively) by three independent readers, as previously described (58). Tissues were graded for peribronchiolar inflammation (around 3–5 small airways per section), and alveolitis in multiple random fields per section. Lung sections were viewed with a Leica DMI3000 B microscope (Leica Microsystems, Inc., Bannockburn, IL), and photographed at 10 × magnification unless otherwise stated, using a Leica DFC 490 camera with the Leica Application software. Immunoperoxidase staining of paraffin-embedded lung sections to examine the distribution of viral antigens was performed as previously described (58).
Assessment of lung edema and vascular leak
The lung wet-to-dry weight ratio was used as an index of lung water accumulation during IAV infection, and was performed as previously described (58). The concentration of protein in BAL supernatants was measured by adding Bradford protein dye. A standard curve using BSA was constructed, and the optical density (OD) determined at 595 nm.
Quantification of inflammatory mediators in bronchoalveolar lavage fluid
The levels of IL-6, GM-CSF, IFN-γ, TNF-α, KC, MCP-1, MIP-1α, MIP-1β, and RANTES in BAL supernatants from virus-infected mice were determined using the mouse cytokine 23-plex Bio-Plex assay (Bio-Rad Laboratories, Inc., Hercules, CA), according to the manufacturer's instructions. Data were collected with a minimum of 100 beads per sample analyzed using Bio-Plex Manager Software. Concentrations were calculated from a standard curve and expressed as picograms per milliliter.
Statistical analysis
For the comparison of two sets of values, a Student's t-test (two-tailed, two-sample equal variance) was used. When comparing three or more sets of values, a one-way analysis of variance (ANOVA) was applied. A p value of ≤0.05 was considered statistically significant.
Results
Influenza viruses infect AEC to similar levels, but differ markedly in their ability to infect alveolar MΦ
In previous reports, we have demonstrated that BJx109 infects murine macrophages efficiently, whereas PR8 does not (44,47,59). We first confirmed these observations in independent experiments using alveolar MΦ, and for comparison, primary mouse AEC. Immunofluorescence confirmed that BJx109 infected alveolar MΦ to high levels (∼90%; Fig. 1A), and HKx31 infected MΦ to intermediate levels (∼60%), whereas PR8 was poor in its ability to infect airway MΦ (∼5–10%). In contrast, virus strains infected primary AEC to equivalent levels (∼70%; Fig. 1A).
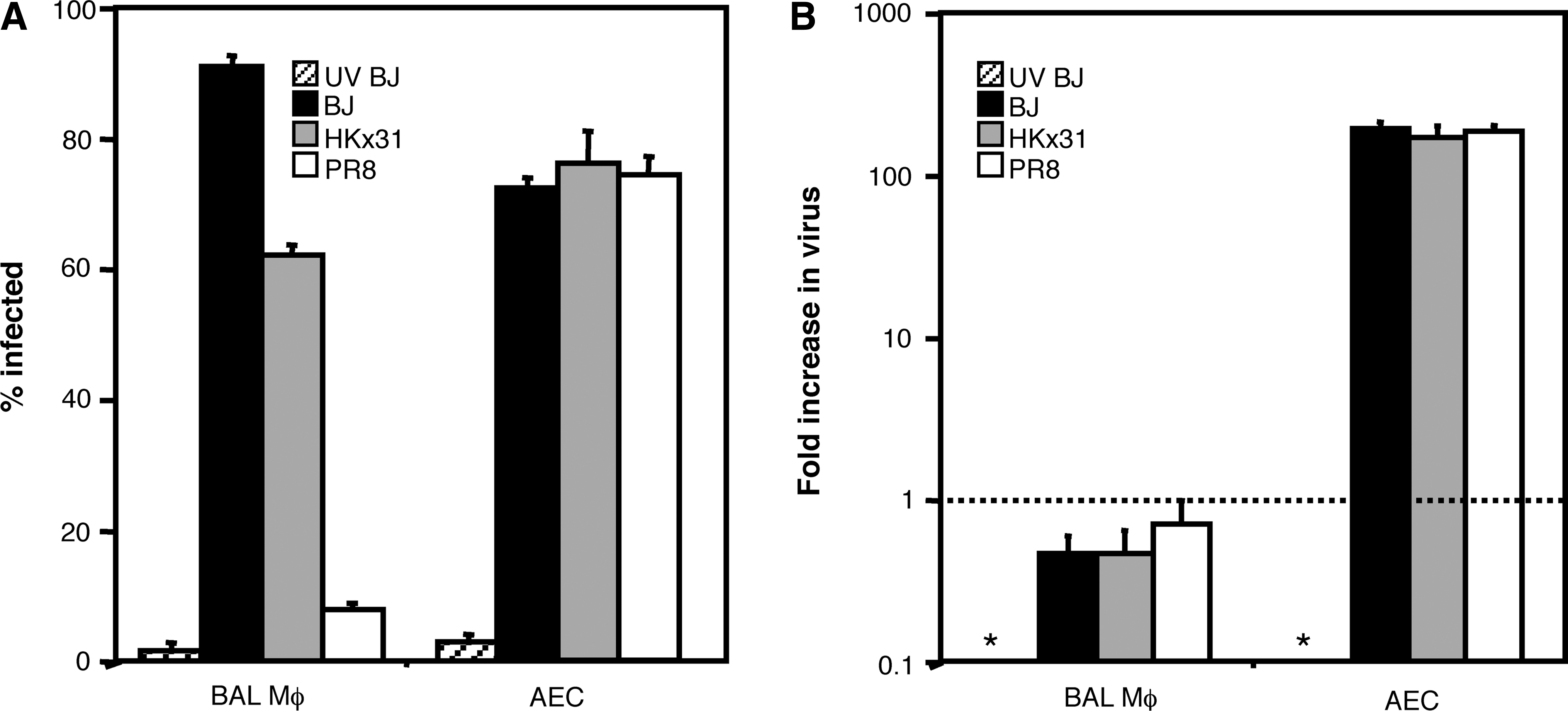
The ability of influenza virus to infect alveolar MΦ and alveolar epithelial cells (AEC) in vitro. Monolayers of bronchoalveolar lavage (BAL) MΦ or primary AEC were incubated with 106 PFU of BJx109 (BJ), HKx31, or PR8, or with an equivalent dose of BJx109 that had been UV-irradiated for 30 min to destroy virus infectivity (UV BJ). After 1 h at 37°C, the monolayers were washed and cultured in serum-free media. (
To assess the ability of IAV to replicate in mouse MΦ and epithelial cells, monolayers of BAL MΦ or AEC were inoculated with 106 PFU of BJx109, HKx31, or PR8, or with an equivalent dose of UV-inactivated BJx109. After 1 h, the cells were washed and fresh serum-free media was added. Supernatants were harvested 2 or 24 h after exposure to virus, and infectious virus present was determined by standard plaque assay on MDCK cells. Data are expressed as the fold increase between 2 and 24 h (Fig. 1B). Incubation of MΦ with each virus strain did not result in virus amplification, consistent with previous reports that influenza A virus infection of murine MΦ is non-productive (48,65). In contrast, infectious virus in supernatants from AEC exposed to BJx109, HKx31, or PR8 increased >100-fold between 2 and 24 h, consistent with the ability of epithelial cells to support productive virus infection (3,46).
Production of inflammatory mediators by alveolar MΦ and AECs following exposure to BJx109, HKx31, or PR8
Next, we determined the spectrum and levels of inflammatory mediators produced following exposure of mouse MΦ or AEC to IAV (Fig. 2). Overall, alveolar MΦ exposed to IAV produced high levels of TNF-α, MIP-1α, RANTES, MIP-2, and KC, and low levels of MCP-1, whereas virus-infected AEC produced high levels of MCP-1, RANTES, MIP-2, and KC, and low levels of TNF-α and IL-6. Thus the overall spectrum of cytokine/chemokine production was different for mouse MΦ and epithelial cells. The viruses differed in their ability to induce cytokine and chemokine secretion from MΦ. BJx109 and HKx31 induced TNF-α, MIP-1α, RANTES, MIP-2, and KC, whereas an equivalent dose of PR8 did not. The induction of all cytokines and chemokines examined was dependent on infectious virus, as inflammatory mediators from MΦ or AEC exposed to UV-irradiated BJx109 were no different from the levels in mock-infected cells.
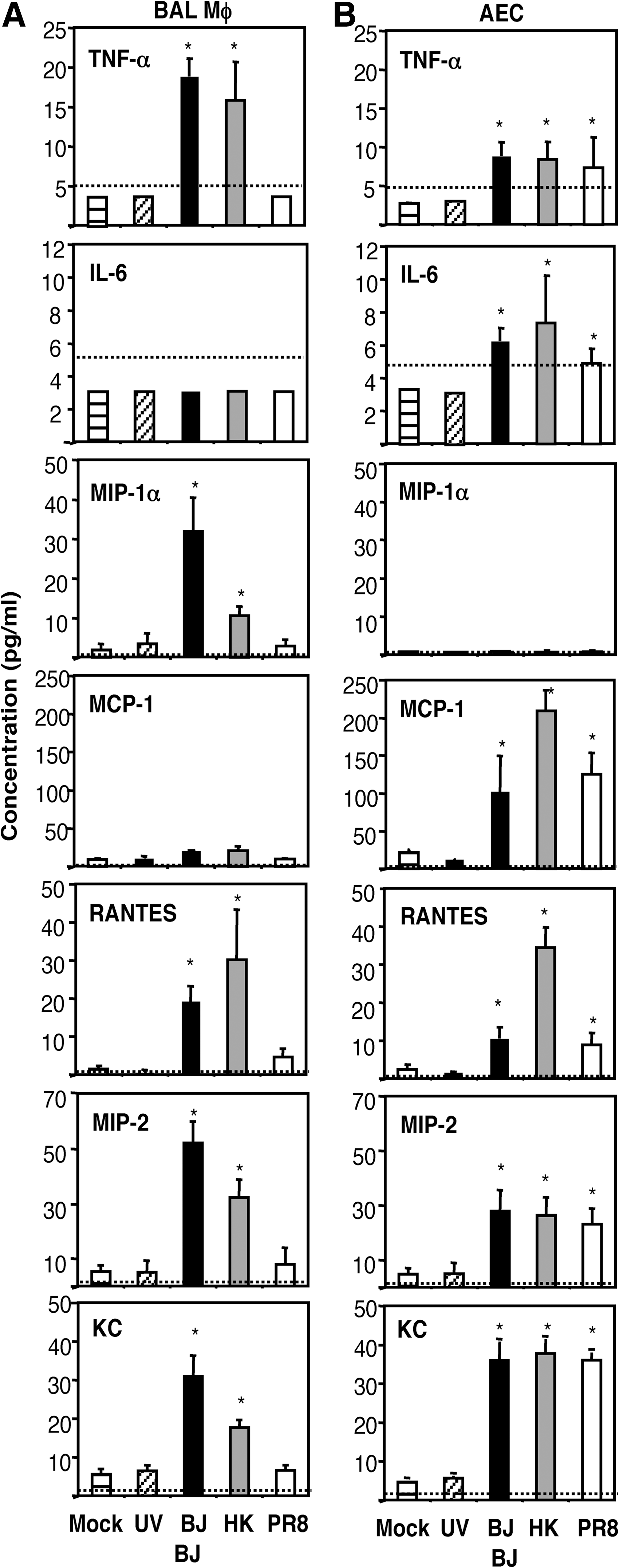
Production of inflammatory mediators by alveolar MΦ and alveolar epithelial cells (AEC) following exposure to influenza viruses. Monolayers of bronchoalveolar lavage (BAL) MΦ and AEC were incubated for 1 h at 37°C with 106 PFU of BJx109 (BJ), HKx31 (HK) or PR8, or with media alone (Mock). Cell monolayers were also incubated with 106 PFU of BJx109 that had been UV-irradiated for 30 min to destroy virus infectivity (UV BJ). The cells were washed and incubated for 24 h, and the supernatants were removed, clarified, and assayed for a range of inflammatory mediators. Cytokine/chemokine concentrations were assayed via ELISA or cytokine bead array (CBA) as described in the Materials and Methods section. Data represent the mean ± 1 SD from three independent experiments. The detection limit of each assay is indicated as a dotted line (TNF-α = 5 pg/mL, IL-6 = 5 pg/mL, MIP-1α = 2 pg/mL, MCP-1 = 5 pg/mL, RANTES = 2 pg/mL, MIP-2 = 2 pg/mL, KC = 2 pg/ml; *levels were significantly different from those in supernatants from mock-infected cells; p < 0.05 by one-way ANOVA).
BJx109, HKx31, and PR8 differ in virulence in C57BL/6 mice
We hypothesized that the tropism of each virus strain for particular cells of the respiratory tract could be an important factor contributing to virulence and disease. Extending our previous studies comparing the virulence of BJx109 and PR8 in mice (59), B6 mice were inoculated via the intranasal route with 105 PFU (Fig. 3A) or 102 PFU (Fig. 3B) of BJx109, HKx31, or PR8, and disease was assessed by weight loss (Fig. 3, upper panels) and survival (Fig. 3, lower panels). Intranasal inoculation with 105 PFU of BJx109 did not induce significant weight loss; however, mice inoculated with an equivalent dose of HKx31 showed progressive weight loss over the first 6–7 d of infection, but all animals regained weight and survived the infection. Mice infected with 105 PFU of PR8 lost weight rapidly and all animals were euthanized. B6 mice infected with 102 PFU of BJx109 or HKx31 did not show significant weight loss (Fig. 3B); however, all mice infected with an equivalent dose of PR8 lost weight progressively and succumbed to disease within 9–10 d. Thus, BJx109, HKx31, and PR8 display clear differences in their ability to induce disease, and may be classified as avirulent, of intermediate virulence, and highly virulent, respectively, in B6 mice.
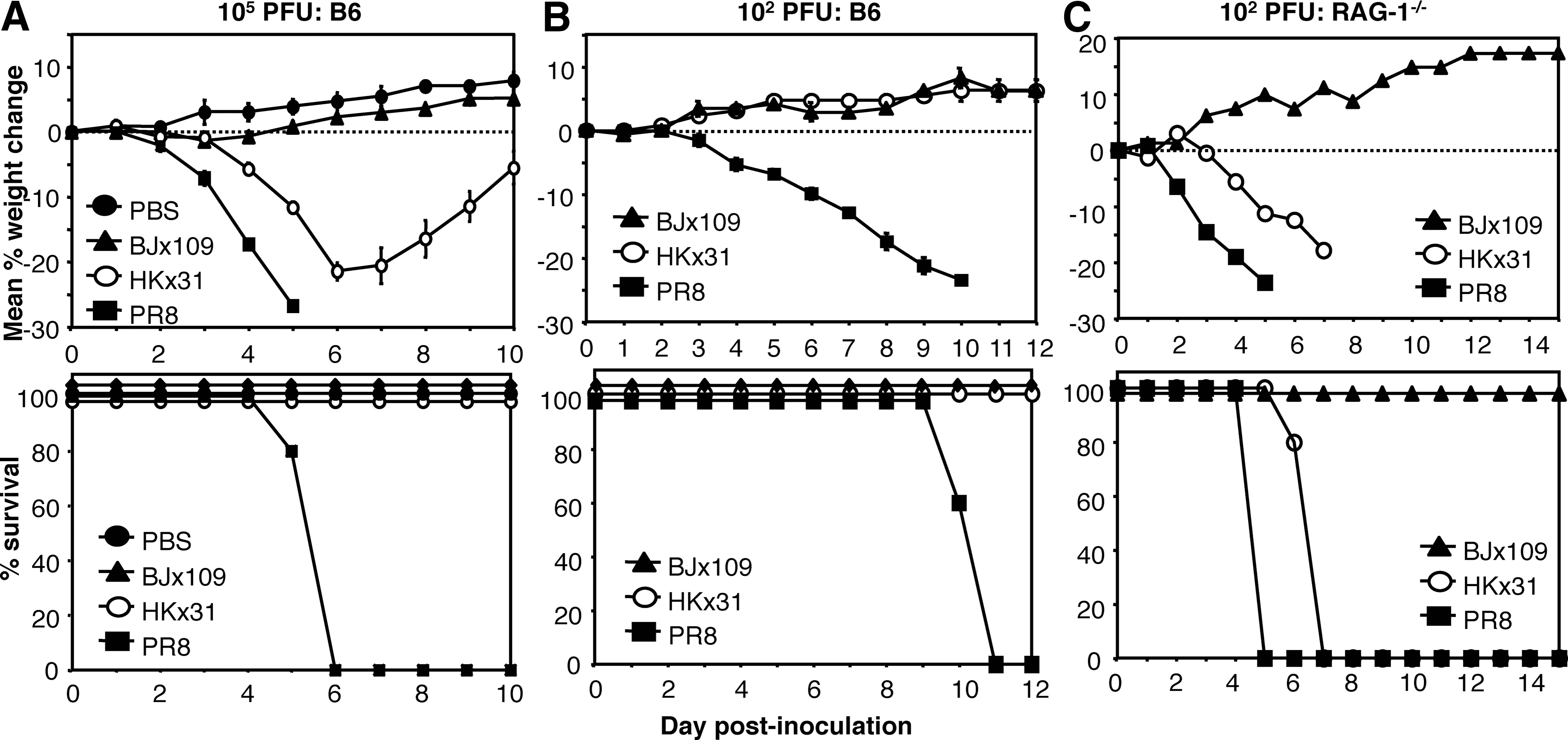
Virulence of influenza strains BJx109, HKx31, and PR8 for B6 mice. Groups of five C57BL/6 (B6) mice were infected via the intranasal route with (
Next, we compared virus strains for their ability to induce disease following IN inoculation of immunodeficient B6.RAG-1−/− mice (37). Inoculation of B6.RAG-1−/− mice with 102 (Fig. 3C) or 105 PFU (data not shown) BJx109 was not associated with significant weight loss or disease over a 15-d monitoring period. Immunocompetent B6 mice inoculated with 102 PFU of HKx31 showed no signs of weight loss (Fig. 3B, upper panel); however, all B6.RAG-1−/− mice inoculated with an equivalent dose lost weight (Fig. 3C, upper panel), and succumbed to disease (Fig. 3C, lower panel). B6 or B6.RAG-1−/− mice inoculated with 102 PFU of PR8 all succumbed to disease (Fig. 3C, lower panel), although PR8-infected B6.RAG-1−/− mice lost weight more rapidly and were euthanized earlier than B6 mice (Fig. 3C, upper panel). Thus, adaptive immunity is not required to restrict the ability of BJx109 to induce disease in B6 mice.
Viral replication and pulmonary inflammation following intranasal infection of B6 mice with BJx109, HKx31, or PR8
B6 mice were infected with 105 PFU of BJx109, HKx31, and PR8, and viral loads in the lower (lungs) and upper (nasal tissues) respiratory tract were examined at days 1–9 post-infection (Fig. 4). Viral titers in the lungs (Fig. 4A) and nasal tissues (Fig. 4B) of mice infected with BJx109 virus peaked 1 d after infection, and were below the level of detection by day 5, consistent with containment of virus via the innate immune system. HKx31 replicated to substantial levels in the lungs at 1–5 d post-infection, but titers were markedly reduced 7–9 d post-infection. Viral titers were high 1 d after PR8 infection, and remained so at 3 and 5 d post-infection, when the mice were euthanized due to excessive weight loss and disease (Fig. 3).
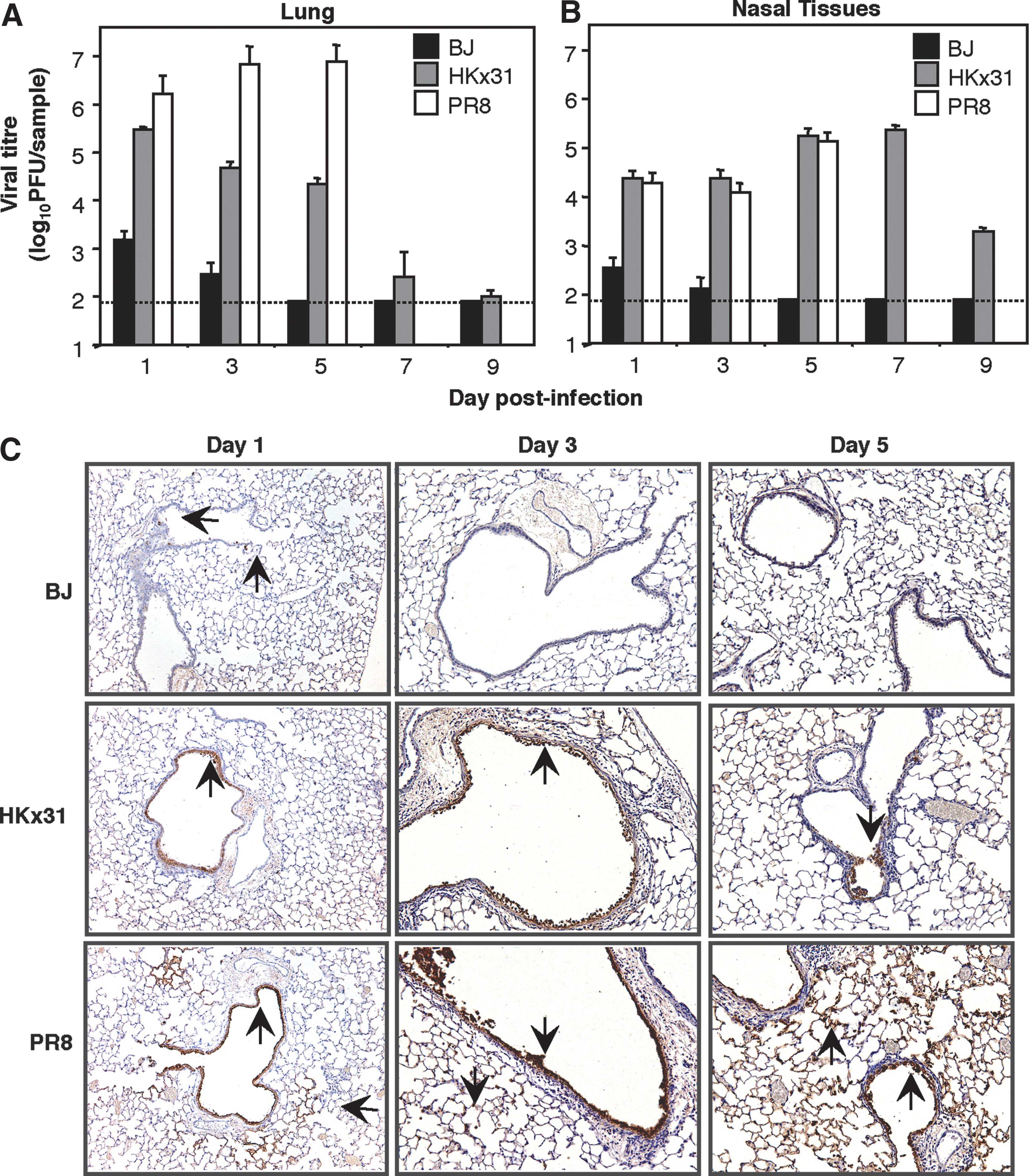
Virus titers in the respiratory tract of B6 mice infected with BJx109 (BJ), HKx31, or PR8. Groups of five B6 mice were infected via the IN route with 105 PFU BJx109, HKx31, and PR8. Mice were euthanized and analyzed at days 1, 3, and 5 post-infection. (
HKx31 and PR8 showed marked differences in their pattern of replication in the lung; however, both viruses replicated to similar levels in nasal tissues at days 1, 3, and 5 post-infection (Fig. 4B). As BJx109, HKx31, and PR8 were equally efficient in their ability to replicate productively in primary AEC (Fig. 1B), differences in virus titers are likely to reflect differences in sensitivity to components of innate host defense. Moreover, we have previously shown that BJx109 can replicate to high titers (>4.0 log10 PFU/lung) in the lungs of mice following depletion of airway MΦ (59), demonstrating that this strain can grow efficiently in the lung in the absence of specific innate defenses.
Immunoperoxidase staining of lung sections from virus-infected animals was performed to determine the distribution of viral antigen 1, 3, and 5 days after IN infection with IAV. Occasional cells staining positive for viral antigen were detected in the lungs of BJx109-infected mice at day 1 post-infection, but not at day 3 or 5 (Fig. 4C, upper panels), consistent with the low titers of infectious virus recovered from the lungs of these animals (Fig. 4A). Following infection with HKx31, viral antigen localized to epithelial cells lining the bronchioles and small airways at 1, 3, and 5 d post-infection (Fig. 4C, middle panels). Epithelial cells lining the bronchioles and small airways of PR8-infected mice stained strongly for viral antigen at days 1, 3, and 5 post-infection (Fig. 4C, lower panels), with increasing involvement of epithelial cells lining the alveolar compartments at the latter time points. By day 5 post-infection, cells stained strongly for viral antigen throughout the airways and alveoli of PR8-infected mice.
The severe clinical disease observed following infection with PR8 was characterized by enhanced recruitment of inflammatory cells to the airways (Fig. 5A), particularly neutrophils, NK cells, T cells, and B cells (Fig. 5B). Moreover, PR8 infection resulted in lung injury, characterized by increased vascular leakage (Fig. 5C), and pulmonary edema (Fig. 5D), compared to naïve animals or to animals infected with BJx109 or HKx31.
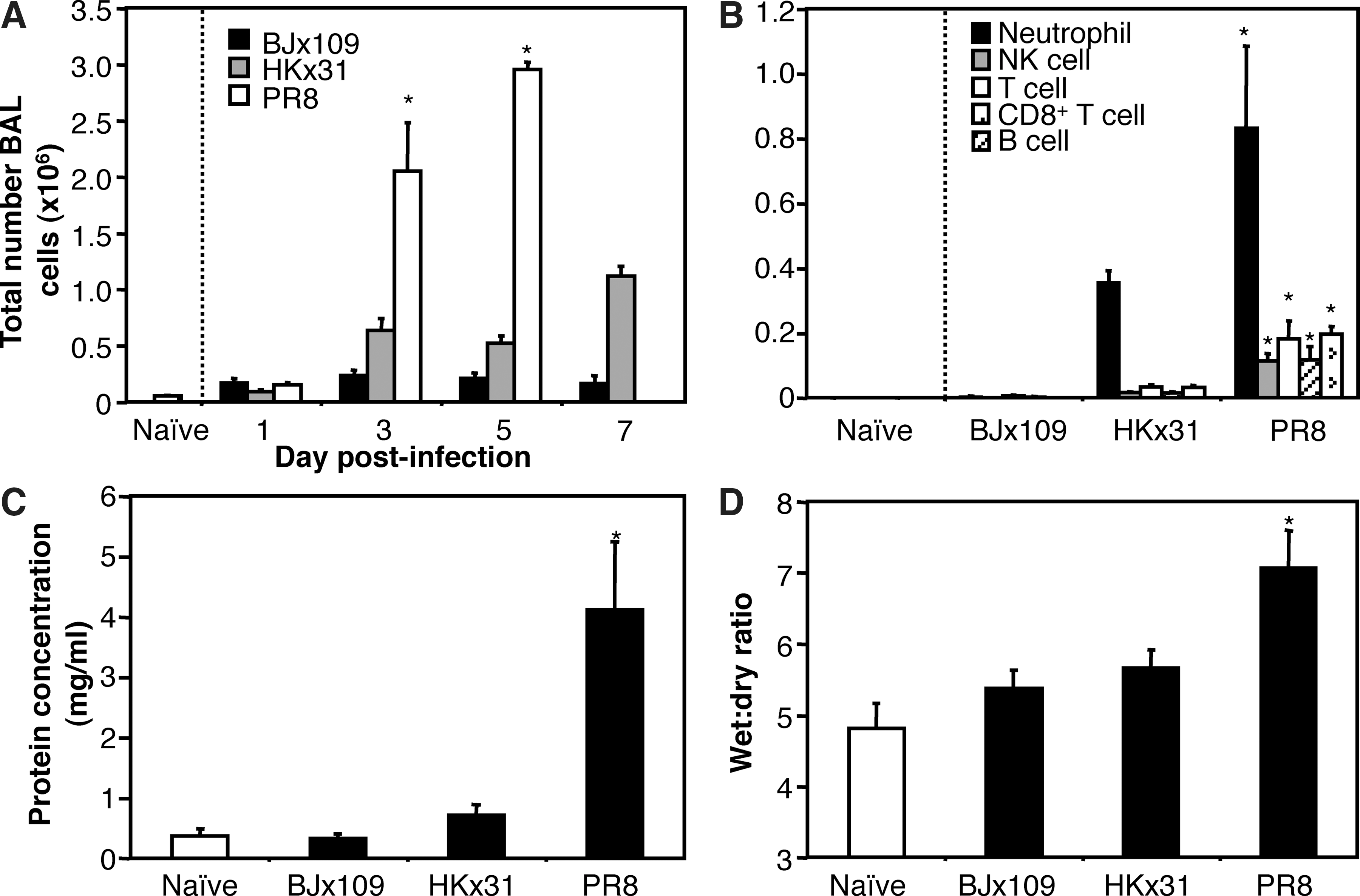
Severe disease during PR8 infection of B6 mice is associated with increased inflammation and pulmonary edema. Groups of five B6 mice were infected with 105 PFU of BJx109, HKx31, or PR8. (
Levels of cytokines and chemokines in BAL supernatants following infection with BJx109, HKx31, and PR8
The magnitude and nature of the cellular infiltrate in the airways and/or the production of local immunomodulators could be critical factors contributing to morbidity and mortality during IAV infection. Therefore we determined levels of cytokines and chemokines in BAL supernatants from virus-infected mice at 1, 3, and 5 days post-infection (Fig. 6). Levels of TNF-α, IL-6, MIP-1α, MCP-1, RANTES, and KC were below the level of detection in BAL supernatants of BJx109-infected mice at all time points tested, although low levels of MIP-2 were recorded at all times. In contrast, HKx31 and PR8 induced significant levels of a range of soluble inflammatory mediators. HKx31 induced production of variable levels of MIP-1α, MCP-1, RANTES, MIP-2, and KC over the course of infection. Infection of mice with PR8 was associated with production of TNF-α, IL-6, MCP-1, KC, and MIP-2. Compared to HKx31, infection of mice with PR8 was associated with additional production of TNF-α and IL-6 during the early phase of infection. Of interest, MIP-1α, produced by airway MΦ but not AEC in response to IAV infection in vitro (Fig. 2), was detected in BAL fluids of HKx31-infected animals, but not PR8-infected animals.
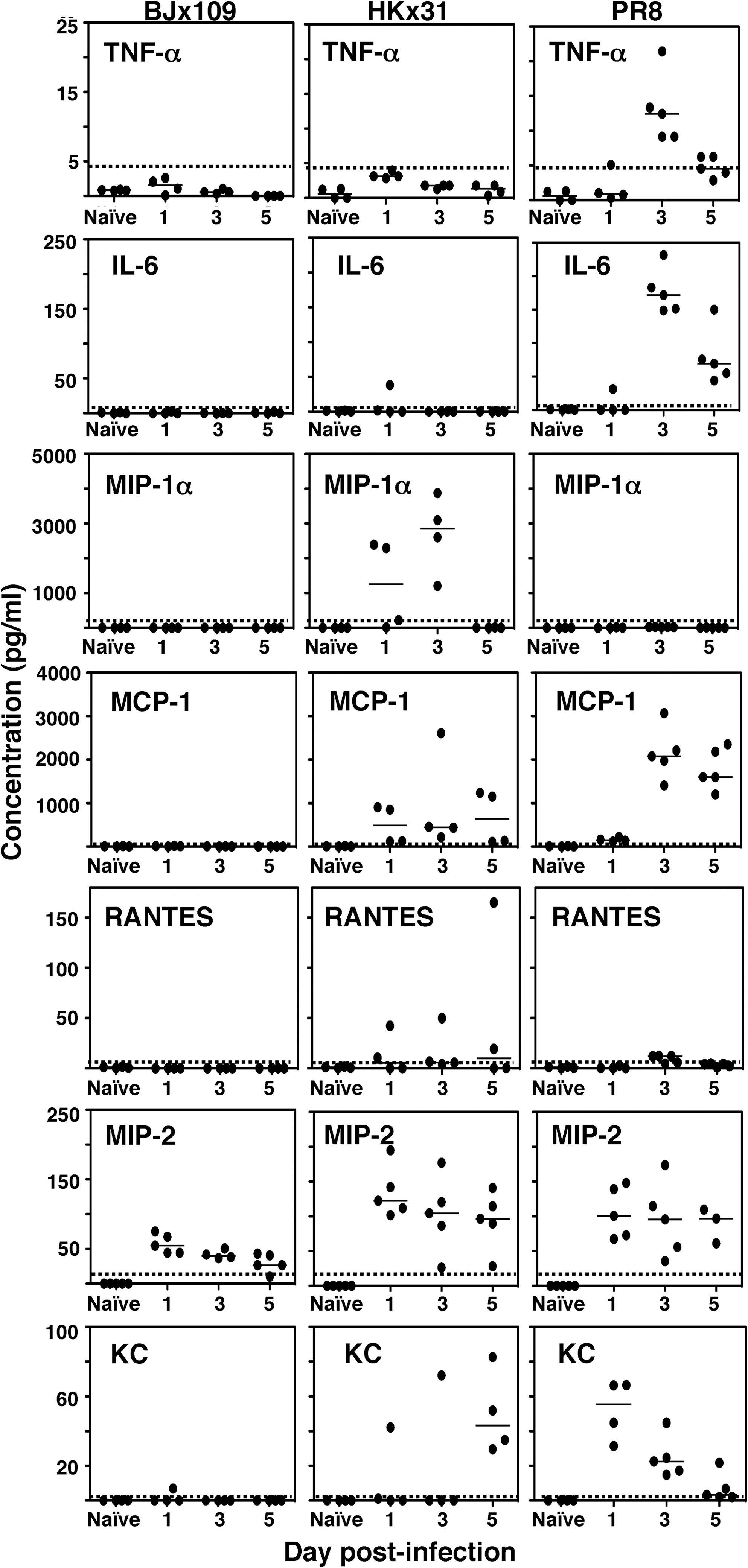
Inflammatory mediators in the airways of mice infected with BJx109, HKx31, or PR8 viruses. Groups of five B6 mice were infected via the IN route with 105 PFU BJx109, HKx31, or PR8. Mice were euthanized at days 1, 3, and 5 post-infection, and the levels of inflammatory mediators in BAL supernatants from naïve or virus-infected mice were determined via Bio-plex assay as described in the Materials and Methods section. Results are expressed in picograms per milliliter; individual mice are indicated by black circles, and the mean is indicated by a horizontal bar. The detection limit for each mediator is indicated by a dotted line (TNF-α = 5 pg/mL, IL-6 = 1 pg/mL, MIP-1α = 5 pg/mL, MCP-1 = 3 pg/mL, RANTES = 0.5 pg/mL, MIP-2 = 5 pg/mL, KC = 3 pg/mL.
Neutralizing activity of mouse airway fluids blocks the ability of influenza viruses to infect MΦ or AEC
BJx109 was efficient in its ability to infect both alveolar MΦ and AEC in vitro (Fig. 1A), yet the virus replicated poorly in mouse lungs (Fig. 4A), and few antigen-positive cells could be detected via immunoperoxidase staining (Fig. 4C). Moreover, few inflammatory mediators were induced following IN infection of mice with BJx109 (Fig. 6). The respiratory tract is lined by surfactant containing a number of proteins of the innate immune system known to mediate antiviral activity against IAV [reviewed in (12)]. Therefore, we compared the sensitivity of BJx109, HKx31, and PR8 to neutralization by cell-free BAL fluids from naïve B6 mice. Consistent with published data (57), pre-incubation of BJx109 with mouse BAL inhibited its ability to infect MDCK epithelial cells, and neutralizing activity was reversed in the presence of mannan (Fig. 7A), indicating that SP-D, rather than SP-A, was the major agent acting against BJx109. In contrast, mouse BAL exhibited weak neutralizing activity against PR8, and this was unaffected by mannan. The sensitivity of HKx31 to neutralization by mouse BAL was intermediate, between that of BJx109 and PR8.
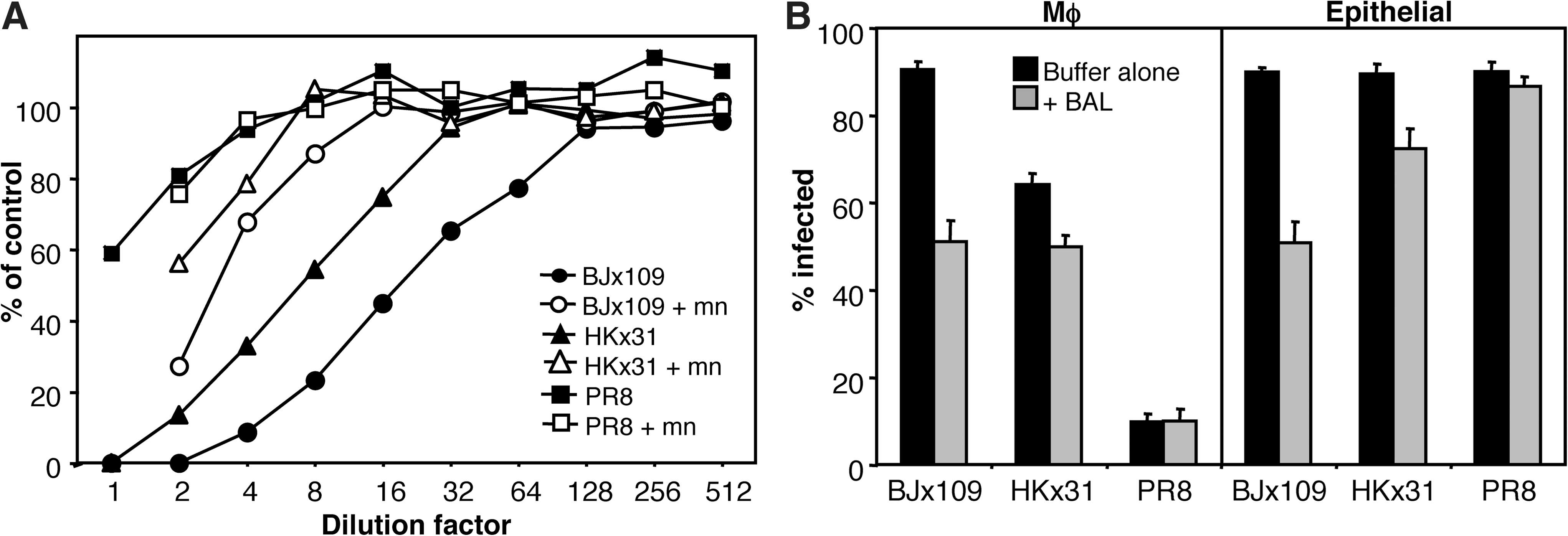
Antiviral activity of mouse BAL fluids against influenza viruses. (
Next, murine MΦ or AEC were pre-incubated with mouse BAL prior to the addition of either BJx109 or PR8. As seen in Fig. 7B, mouse BAL blocked infection of MΦ and AEC by BJx109, but was markedly less efficient against PR8. Again, the inhibitory activity of mouse BAL in blocking infection of MΦ and AEC by HKx31 was intermediate compared to BJx109 and PR8. Thus, mannose-binding lectins present in fluids lining the murine respiratory tract protect alveolar MΦ and AEC from infection with BJx109, but not PR8. These data are consistent with our findings that BJx109 induces high levels of chemokines and cytokines from mouse airway cells in vitro (Fig. 2), but not in vivo, following IN infection of mice (Fig. 6).
Discussion
Inflammatory mediators produced by AEC and alveolar MΦ orchestrate the cell-mediated inflammatory response elicited following respiratory infection with IAV. Moreover, the tropism of different virus strains for epithelial cells and/or MΦ is likely to determine the spectrum of chemokines and cytokines produced. In this study, BJx109, HKx31, and PR8 were shown to differ in virulence in mice, and were classified as avirulent, of intermediate virulence, and highly virulent, respectively. As BJx109 and HKx31 bear internal components derived from PR8, these differences in virulence must relate to properties of the HA/NA surface glycoproteins rather than internal components of the virion. Based on these findings, we investigated factors that contribute to the virulence of PR8 in mice. In particular, PR8 did not elicit innate responses from airway MΦ in vitro, and was resistant to neutralization by mouse airway fluids. Moreover, PR8 was shown to replicate and spread throughout the lung, inducing severe viral pneumonia. Together these data indicate that the ability of PR8 to evade components of innate immune defense is likely to be an important factor contributing to its virulence in mice.
The ability of BJx109, HKx31, and PR8 to infect airway MΦ correlated with their ability to induce inflammatory mediators from these cells. Accordingly, BJx109 was a potent inducer of inflammatory mediators (TNF-α, MIP-1α, RANTES, MIP-2, and KC) from airway MΦ, whereas PR8 was not (Fig. 2). Mouse airway MΦ produce TNF-α, but not MCP-1, in response to IAV (24), and IAV-infected airway MΦ from pigs produced IL-1β, IL-6, IL-8, and TNF-α (51). however, most in-vitro studies have utilized human monocytes/MΦ. In contrast to mouse MΦ, human MΦ exposed to PR8 secreted a range of inflammatory mediators, including IL-1β, IL-6, TNF-α, MIP-1α, MCP-1, and RANTES (7,17,25,43,54). Sialic acid is the primary attachment receptor for IAV on target cells [reviewed by (52)], and human airway MΦ express predominantly α(2,3)-Gal-linked sialic acid (39,40). In contrast, we have found α(2,6)-Gal-linked sialic acid to be the major linkage expressed by mouse airway MΦ (56). The preference of PR8 for α(2,3)-Gal-linked sialic acid (55,62) is consistent with its ability to infect human, but not mouse, MΦ. Furthermore, in contrast to mouse MΦ, some human MΦ have been reported to support productive IAV infection (42), suggesting that human and mouse MΦ differ in their ability to recognize and respond to IAV.
Mouse AEC exposed to BJx109, HKx31, or PR8 produced MCP-1, RANTES, MIP-2, and KC, as well as some TNF-α and IL-6 (Fig. 2). These findings are consistent with previous reports of IAV-induced production of TNF-α, IL-6, and type I IFN from murine tracheal epithelial cells (38), and PR8-induced secretion of MCP-1 and RANTES from mouse AEC (24). Human AEC exposed to IAV produce RANTES, MCP-1, and IL-8 (2,29), as well as IP-10, IL-6, and IFN-β (8 –10). Based on in vitro studies, it has been proposed that human AEC secrete only neutrophil-attracting chemokines (such as IL-8) in response to IAV, whereas human MΦ secrete mononuclear leukocyte-attracting chemokines, and actually suppress neutrophil-attracting chemoattractants (2,29,53). KC/MIP-2, functional homologues of human IL-8 (5,60), were produced by mouse AEC in response to IAV (Fig. 2), and by mouse airway MΦ in response to BJx109 and HKx31 (Fig. 2). Thus, mouse airway cells (AEC and MΦ) do not display the polarized cytokine/chemokine profiles reported following exposure of human cells to IAV.
To better understand influenza-induced disease in mice, we characterized IAV-induced cytokine and chemokine production in vivo. BJx109, HKx31, and PR8 induced mild, moderate, and severe disease, respectively (Fig. 3). In vitro, BJx109, HKx31, and PR8 infected AEC to equivalent levels (Fig. 1A), yet in vivo BJx109 replicated poorly, and few IAV-positive cells were detected in the lung (Fig. 4C). Thus caution must be exercised in extrapolating in vitro observations to the complex microenvironment of the lung. In vivo, respiratory epithelium is lined by pulmonary surfactant, a rich source of proteins of the innate immune system that mediate antiviral activity against IAV (19,22,45). Mouse BAL inhibited the ability of BJx109, but not PR8, to infect mouse AEC and MΦ (Fig. 7B), a finding consistent with reports that PR8 is resistant to neutralization by SP-D (20,45). These results suggest that BJx109 is neutralized by SP-D in lung fluids before it can infect the respiratory epithelium, and are consistent with our findings that BJx109 induced high levels of inflammatory mediators from airway MΦ in vitro, yet levels in BAL were very low or below the level of detection.
Infection of mice with PR8 led to rapid weight loss and disease (Fig. 3), associated with extensive virus replication and spread in the lungs (Fig. 4), and pulmonary inflammation and damage to lung epithelium (Fig. 5). Several independent studies have reported a rise in the levels of inflammatory mediators (e.g., TNF-α, IL-1α, IL-1β, IL-6, and MCP-1) in BAL or lung homogenates in temporal association with the development of symptoms and lung pathology (23,32,41). In contrast to the protective responses elicited in BJx109- and HKx31-infected mice, PR8 infection was associated with progressive accumulation of leukocytes, leading to lung inflammation and pulmonary edema (Fig. 5). These hallmark features of acute respiratory distress syndrome (ARDS) have previously been associated with severe disease in mice infected with H5N1 (67) or H9N2 (14) viruses, and in influenza virus–infected mice in which specific components of the immune response have been depleted (58,59). The failure of PR8 to elicit innate responses from alveolar MΦ, and its resistance to mouse BAL, favor immediate infection and replication in AEC in the early phase of infection. Once PR8 infection is established, continued replication and amplification of virus results in tissue damage; the high levels of chemokines and cytokines that we observed in BAL from PR8-infected mice (Fig. 6) are consistent with previous findings (6,23,64). BJx109, HKx31, and PR8 did not differ in their ability to replicate in mouse AEC in vitro (Fig. 1B), indicating that the different virus loads observed in mouse lungs (Fig. 4A) reflect differences in susceptibility to innate defenses rather than intrinsic differences in the ability to infect and replicate in AEC. The poor ability of PR8 to infect airway MΦ, and its weak sensitivity to innate proteins in BAL, are consistent with high levels of virus growth in mouse lungs.
BJx109, HKx31, and PR8 share internal components derived from PR8, but differ in HA/NA, and thus in sensitivity to components of innate host defense. BJx109 grows poorly in the airways of immunocompetent mice, but can replicate to high titers in mice depleted of airway MΦ (59). PR8 grows to high titers in mouse lungs, is unaffected by depletion of airway MΦ (59), and is resistant to neutralization by mouse BAL (Fig. 7) (57). BJx109 and HKx31 elicited similar levels of cytokines and chemokines from airway MΦ in vitro (Fig. 2), yet BJx109 was markedly more sensitive to inhibition by airway fluids (Fig. 7). The differential sensitivity of BJx109 and HKx31 to inhibitory proteins in the airways might be an important factor contributing to their differential virulence, particularly in B6.RAG-1−/− mice lacking T-cell− and B-cell−mediated immunity.
Experiments to delineate the relative contribution of SP-D and airway MΦ to innate defense against IAV are complicated, as their production and regulation in the airways are intimately related. For example, impaired surfactant catabolism by airway MΦ can result in alveolar proteinosis [reviewed in (11,26)]. Moreover, researchers have used knockout mice to demonstrate the importance of SP-D in limiting replication of highly glycosylated IAV (22,34,63); however, interpretation is complicated by the aberrant pulmonary homeostasis in SP-D-deficient mice, which includes MΦ activation and surfactant phospholipid accumulations (27,28,31). In our studies, infection of mouse MΦ and epithelial cells was also modulated in the presence of BAL (Fig. 7). The intimate association of surfactant and airway MΦ therefore appears important in mediating coordinated control of IAV by the innate immune system. SP-D in surfactant and MMR on airway MΦ are soluble and cell-associated C-type lectins that recognize glycans on HA/NA to destroy IAV. The ability of PR8 to avoid detection by C-type lectins of the innate immune system is likely an important factor contributing to its virulence.
Footnotes
Acknowledgments
This study was supported by Project Grant no. 509230 from The National Health and Medical Research Council (NH&MRC) of Australia. P.C.R. is an NH&MRC R.D. Wright Research Fellow. The Melbourne WHO Collaborating Centre for Reference and Research on Influenza is supported by the Australian Government Department of Health and Ageing.
Author Disclosure Statement
M.D.T. carried out the majority of experiments described in this study, analyzed and interpreted data, and prepared figures for the manuscript. H.C.S. prepared the primary mouse airway epithelial cell cultures used in this study. P.C.R. and M.D.T. designed the study, and P.C.R. wrote the manuscript. A.G.B. contributed to interpretation of the data and the writing of the manuscript. All authors read and approved the final manuscript. The authors declare that no conflicting financial interests exist.