
Abstract
Griscelli syndrome type 2 (GS2) is a rare autosomal-recessive disorder associated with a RAB27A gene mutation, and clinically manifesting as hypopigmentation, disseminated chronic encephalitis, and severe immunological disorders characterized by an accelerated hematological phase, also referred to as hemophagocytic syndrome (HS), or hemophagocytic lymphohistiocytosis (HLH). The authors report the diagnosis of GS2 in an 11-year-old girl with hypopigmentation, immunodeficiency, hepatosplenomegaly, severe neurological impairments, and fatal multiorgan failure. In this patient a diagnosis of pulmonary lymphomatoid granulomatosis (LG), an Epstein-Barr virus (EBV)-related lymphoproliferative disorder, was established from radiological and histological findings. Although EBV-related malignancies are common in immunocompromised patients, this is the first report of a diagnosis of pulmonary LG in a patient with GS2.
Case Presentation
She was the first child of middle-aged, non-consanguineous parents, born from the second pregnancy, which was terminated in the 38th week of gestation by operative delivery because of fetal acute life-threatening symptoms; after birth she presented with neonatal asphyxia. The perinatal period was complicated by transient tachypnea of the newborn. Until the ninth year of age the child's development was normal; she did not suffer from any serious infectious diseases.
At the age of 9 y she suffered from an upper respiratory tract infection, and afterwards she developed neurological symptoms and features of chronic inflammation of the encephalon on magnetic resonance imaging (MRI) of the central nervous system (CNS). A chest x-ray and high-resolution computed tomography (HRCT) revealed disseminated bilateral interstitial infiltrations in the lungs. Polymerase chain reaction (PCR) examinations excluded: Pneumocystis jiroveci, Candida albicans, cytomegalovirus, and Toxoplasma gondii infection, whereas Epstein-Barr virus (EBV)-DNA was positive. Serological tests for Mycoplasma and Chlamydophila pneumoniae infection were negative. Sputum and blood cultures did not detect either bacterial or mycotic flora. Pancytopenia, coagulopathy, increased activity of aminotransferase enzymes, hypertriglyceridemia, hypofibrinogenemia, and hyperferritinemia, characteristic for hemophagocytic syndrome (HS), were found in laboratory tests. In the face of progressive pancytopenia, bone marrow aspiration and trephine biopsy were performed, revealing features of hemophagocytic lymphohistiocytosis (HLH).
The preliminary diagnosis of Griscelli syndrome type 2 (GS2) based on the patient's phenotype with hypopigmentation and involvement of the CNS, accompanied by abnormalities in immunological and biochemical tests, as well as microscopic examination of child's hair showing large irregular pigment aggregates in the hair shaft, was subsequently confirmed by Rab27a gene sequencing, which identified a heterozygous mutation.
Radiological presentation of the lungs in the next HRCT examination, showing well-confined nodules localized subpleurally and peripherally (Fig. 1), was the indication for an open lung biopsy. Histological examination of lung parenchyma revealed abundant lymphoid infiltrates, consisting of lymphocytes, macrophages, immunoblasts, and disseminated large lymphoid cells involving the bronchial walls and vessels of varying caliber. In the lung parenchyma surrounding these infiltrations, numerous bright, frothy macrophages filling up the lumen of the alveoli were present. This histological pattern corresponded with lymphomatoid granulomatosis grade III (LGIII).
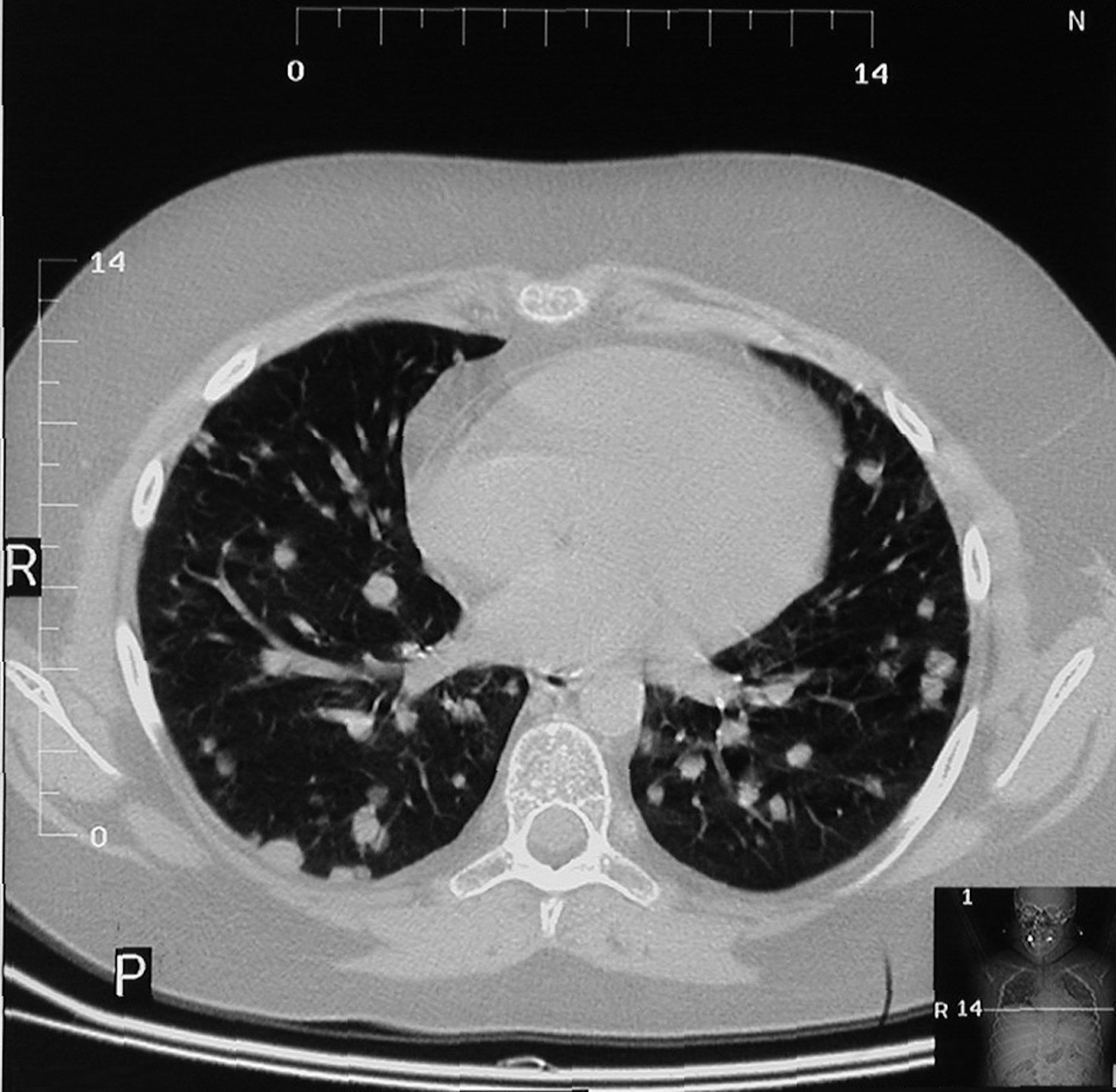
Nodular pattern on high-resolution computed tomography in a patient with Griscelli syndrome type 2 and pulmonary lymphomatoid granulomatosis.
Despite immunosuppressive treatment, development of the next phase, an accelerated hematological phase resistant to therapy, was seen, which rendered the planned hematopoietic stem cell transplant impossible, and led to patient's death because of massive hemorrhage into the CNS.
Discussion
Rab27a guanosine triphosphatase (GTPase) is required for transport of melanosomes and for exocytosis of cytotoxic granules (1,2), therefore its gene mutation leads to hypopigmentation, impaired activity of cytotoxic T lymphocytes, and an immunological disorder presenting as HS (3,4). HS is usually triggered by viral or bacterial infections, and in particular reactivation of a latent EBV infection is often involved in the pathogenesis of this condition (5,6). It has been suggested that EBV infection of B lymphocytes triggers a polyclonal proliferation of cytotoxic T lymphocytes, which in turn stimulate histiocytes and macrophages, resulting in uncontrolled immune activation and subsequent hypercytokinemia, tissue necrosis, and organ insufficiency. Lymphomatoid granulomatosis is a necrotic angiocentric and angiodestructive process in which EBV-positive B-cell proliferation associated with a reactive T-cell proliferation occurs (7). Grade III LG shows numerous, usually monoclonal large EBV-positive B cells, reflecting progressive transformation. This condition should be considered in adult patients presenting with multiple pulmonary nodules (8). A pulmonary manifestation is unusual in individuals with GS2. In only one publication (9) did the authors report recurrent pneumonias since early infancy, accompanied by streaky opacities in the upper lung zones on chest x-ray. Pulmonary LG has not been previously diagnosed in patients with GS2.
Footnotes
Acknowledgment
Special acknowledgment is due to Professor Genevieve de Saint-Basile, from Unite de Recherches sur le Developpement Normal et Pathologique du Systeme Immunitaire, Institut National de la Sante et de la Recherche Medicale, Hopital Necker, Paris Cedex, France, for her valuable help and significant contribution to establishing the diagnosis of Griscelli syndrome by mutation analysis.
Author Disclosure Statement
No competing financial interests exist.