
Abstract
Despite the fact that murine cells are not permissive for human immunodeficiency virus type 1 (HIV-1) infection, several investigators have constructed transgenic (Tg) mice to model HIV-1-induced diseases to overcome this restriction. The generation of Tg mice expressing selected HIV-1 genes revealed that Nef harbors a major disease determinant. HIV-1 Nef protein is a molecular adapter able to interact with several cellular partners, interfering with cellular functions. The phenotype of Nef Tg mice was extensively characterized regarding in vivo development of AIDS-like disease and the effects of Nef expression in T lymphocytes, but the functions eventually corrupted by Nef in monocytes and macrophages were less studied. Nef treatment of human monocyte-derived macrophages induces the internalization of the protein and modulates the production and secretion of different chemokines and cytokines by activating specific intracellular signaling pathways (i.e., NF-κB, MAPK, and IRF3). Therefore we set up an in vitro murine macrophage-based model using stabilized cell lines and primary peritoneal macrophages, and treated them with recombinant myristoylated NefSF2 (recNef). Like human cells, murine macrophages responded to Nef treatment, activating IKK-α and IKK-β, JNK, and p38 MAP kinases. Activation of the NF-κB pathway is mandatory for the synthesis and release of a pool of cytokines and chemokines, including IFN-β, that induce tyrosine phosphorylation of the signal transducer and activator of transcription (STAT)-1, STAT-2, and STAT-3, in an autocrine and paracrine manner, confirming that murine macrophages respond to Nef similarly to human ones. These data extend the results previously obtained in human primary macrophages, allowing the use of murine cells in culture to study signaling events modulated by Nef in myeloid-derived cells. In particular, it may be feasible to use macrophages derived from mice knocked out in specific signaling intermediates to obtain greater insight into the mechanism of Nef-induced effects.
Introduction
In T lymphocytes, Nef expression triggers a transcriptional program imitating single-signal T-cell activation, thereby lowering the T-cell receptor (TCR) activation threshold (8). This is essentially achieved through interactions with key molecules involved in TCR signal transduction pathways, as TCR ζ chain, src kinase Lck, and p95Vav (9 –11). In monocytes and macrophages Nef treatment or its intracellular expression induces the synthesis and the release of inflammatory cytokines and/or lymphocytic-attractant chemokines (i.e., macrophage inflammatory protein-1α [MIP-1α]/CCL3 and MIP-1β/CCL4) (12,13). These features appear due to the ability of the viral protein to intercept CD40 signaling, thereby inducing nuclear factor-κB (NF-κB) signal cascade (14). All of these effects create the perfect environment for viral replication. Interestingly, Nef has also been found inside uninfected B cells of lymphoid follicles from infected individuals (15), and recent experimental evidence has shown that it can be transferred to uninfected cells from infected ones via cellular protrusions and/or exosomes (16 –18), thus opening a new road to deepen our insights into the roles of this multifunctional protein (19,20).
Most animal work in HIV pathogenesis studies has been performed using primate animal models (21,22), but many aspects of HIV-1 infection can be approached in transgenic or humanized mice models, even though murine cells and mice cannot be directly infected with HIV-1 (23). Different groups reported that HIV-1 transgenic mice develop an AIDS-related syndrome (4,24). In transgenic mice expressing Nef, thymocytes as well as T lymphocyte functions have been extensively characterized, and it was observed that thymocytes showed a constitutive increase of tyrosine phosphorylation of several substrates including LAT, compared to thymocytes of non-transgenic mice. In addition, upon anti-CD3 stimulation, levels of tyrosine phosphorylation of several substrates were higher in Nef transgenic than in control non-transgenic mice (3). We previously reported that recombinant Nef treatment of human monocyte-derived macrophages (MDMs) allows the rapid activation of NF-κB, members of the mitogen-activated protein kinases (MAPKs, i.e. ERK1/2, JNK, and p38), and interferon regulatory factor-3 (IRF-3) (25), leading to the synthesis and release of inflammatory chemokines and cytokines, as well as IFN-β, that in turn are able to induce the tyrosine phosphorylation of signal transducers and activators of transcription (i.e., STAT1, STAT2, and STAT3). We also reported that STATs phosphorylation is transiently achieved even in MDMs infected with Δenv nef-expressing HIV-1 VSV-G pseudotypes, but not with Δenv/Δnef viral particles (25 –27).
In the present study we have characterized the response of murine macrophages to treatment with recombinant wild-type NefSF2 or mutant protein, in which a series of conserved functional amino acid residues were tested. The results reported here demonstrate that the RAW264.7 murine cell line and primary peritoneal macrophages respond to Nef treatment similarly to human MDMs, rendering murine macrophages from wild-type or transgenic mice a feasible model to study the mechanism of the effects induced both by Nef treatment and expression.
Materials and Methods
Cell cultures and reagents
The RAW264.7 cell line was established from a tumor induced by Abelson murine leukemia virus (28). The J774A.1 cell line was derived from a murine histiocytic lymphoma (29). Both cell lines were grown in RPMI medium supplemented with 10% heat inactivated fetal calf serum (iFCS), penicillin (100 U/mL), streptomycin (100 μg/mL), and glutamine (2 μM), all purchased from Lonza (Milan, Italy).
The following inhibitors were used: the MEK-1 allosteric inhibitor PD98059 (2′-amino-3′-methoxyflavone), which specifically prevents the activation of ERK1/2 MAP kinases (30); the p38-specific inhibitor SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole], which competes with ATP at its binding site (31); and the JNK-specific inhibitor SP600125, which is a reversible competitor of the ATP-binding site (32). All these inhibitors were purchased from Merck Biosciences-Calbiochem (Nottingham, U.K.), and used at a concentration of 10 μM. The highly-specific IKK-α/β inhibitor BMS-345541 (33) was a kind gift from James R. Burke, Department of Immunology, Inflammation and Pulmonary Drug Discovery, Bristol-Myers Squibb Pharmaceutical Research Institute, Princeton, NJ, and was used at a concentration of 25 μM. Recombinant murine interferon-β (IFN-β) was from PBL Interferon Source (Piscataway, NJ). Lipopolysaccharide (LPS; Sigma-Aldrich, Milan, Italy) was purified from the 0111:B4 E. coli strain.
Recombinant Nef protein preparations
Recombinant Nef proteins were obtained as hexahistidine-tagged fusion proteins as previously described (34). In particular, for the development of a myristoylated Nef expression system in E. coli, a codon-optimized Nef gene was amplified by polymerase chain reaction (PCR) with NcoI and HindIII restriction sites at the 5′ and 3′ ends, respectively, and omitted for a stop codon at the 3′ end. This fragment was cloned into the pET-23d expression vector that contained an ampicillin resistance and a C-terminal His6 tag for affinity chromatography. Therefore, upon expression of Nef, residues KLAAALEHHHHHH were attached at the C-terminus to the protein product. Myristoylated and non-myristoylated recombinant Nef mutant proteins were prepared as previously described (35,36). All Nef protein preparations were analyzed for the presence of endotoxin using the chromogenic Limulus amebocyte lysate end-point assay QCL-1000, and if required, purified using the EndoTrap endotoxin removal system (both from Cambrex, Milan, Italy). To avoid possible signaling effects due to residual LPS traces in Nef preparations, all of the experiments were performed in the presence of 1 μg/mL of polymyxin B (Sigma-Aldrich), a cationic antibiotic that binds to the lipid A portion of bacterial LPS. This polymyxin B treatment did not interfere with the signaling events analyzed, and it blocked the signaling activity of up to 50 endotoxin units (EU)/mL LPS.
Western Blot
Cells were washed twice with phosphate-buffered saline (PBS; pH 7.4), and lysed in 20 mM HEPES (pH 7.9), 50 mM NaCl, 10 mM EDTA, 2 mM EGTA, 0.5% nonionic detergent IGEPAL CA-630 (Sigma-Aldrich), 0.5 mM dithiothreitol (DTT), 20 mM sodium molybdate, 10 mM sodium orthovanadate, 100 mM sodium fluoride, 10 μg/mL leupeptin, and 0.5 mM phenylmethylsulfonylfluoride for 20 min in ice. Whole-cell lysates were centrifuged at 6000 g for 10 min at 4°C, and the supernatants were frozen at 80°C. The protein concentrations of cell extracts were determined by Lowry's protein assay. Aliquots of cell extracts containing 20–50 μg of total proteins were resolved on 7–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to nitrocellulose membranes (Sartorius, Göttingen, Germany) by electroblot for 60 min at 100 V with a Bio-Rad Trans-Blot (Bio-Rad, Hercules, CA). For the immunoassay, the membranes were blocked in 3% bovine serum albumin (BSA) fraction V (Sigma-Aldrich) in TTBS/EDTA (10 mM Tris, pH 7.4; 100 mM NaCl; 1 mM EDTA; and 0.1% Tween 20) for 1 h at room temperature, and then incubated for 1 h at room temperature or overnight at 4°C with specific antibodies diluted in 1% BSA/TTBS-EDTA. The antibodies used in the different immunoblots were as follows: rabbit polyclonal antibodies anti-phospho-IKK-α(Ser180)/IKKβ(Ser181), anti-Iκ-B-α, anti-phospho-STAT1(Tyr701), anti-phospho-STAT3(Tyr705), anti-phospho-SAPK/JNK(Thr183/Tyr185), anti-SAPK/JNK, anti-phospho-p38(Thr180/Tyr182), and anti-p38 MAP kinase, all from Cell Signaling Technology (Beverly, MA). Anti-phospho-Stat2(Tyr689) was from Millipore (Billerica, MA); rabbit polyclonal antibodies anti-STAT1 (E-23), anti-STAT2 (L-20), anti-STAT3 (C-20), anti-IRF-1 (C-20), anti-IRF-3 (FL-425), and anti-TFIIH p62 (Q-19), were all from Santa Cruz Biotechnology (Santa Cruz, CA); mouse monoclonal anti β-tubulin was from ICN Biomedicals (Costa Mesa, CA). Immune complexes were detected with horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse antiserum (Calbiochem, San Diego, CA), followed by enhanced chemiluminescence reaction (ECL; Amersham Pharmacia Biotech, Milan, Italy). To reprobe membranes with antibodies having different specificities, nitrocellulose membranes were stripped for 5 min at room temperature with Restore Western Blot Stripping Buffer (Pierce, Rockford, IL), and then extensively washed with TTBS/EDTA.
Nuclear and cytoplasmic extract preparation for IRF-3 nuclear translocation analysis
RAW264.7 (2×106 cells) were treated with myr+ recNefSF2, harvested in ice-cold PBS buffer, and then centrifuged at 1200 rpm for 5 min at 4°C. Cell pellets were lysed with 200 μL of hypotonic buffer (10 mM HEPES, pH 7.8; 10 mM KCl; 1 mM MgCl2; 0.1 mM EGTA; 1 mM EDTA; 5% glycerol; 1 mM PMSF; 1 μg/mL pepstatin A; 2.3 μg/mL aprotinin; 1 μg/mL leupeptin; 1 mM Na3VO4; 22 mM NaF), and incubated on ice for 15 min. Then 0.58% IGEPAL CA-630 (Sigma-Aldrich) was added, the samples were incubated on ice for 2 min, and were centrifuged at 14,000 rpm for 5 min. Supernatants corresponding to the cytoplasmic fraction were harvested, whereas 60 μL of hypertonic buffer (50 mM HEPES, pH 7.8; 400 mM NaCl; 1 mM MgCl2; 1 mM EGTA; 1 mM EDTA; 10% glycerol; 1 mM PMSF; 1 μg/mL pesptatin A; 2.3 μg/mL aprotinin; 1 μg/ml leupeptin; 1 mM Na3VO4; 22 mM NaF) were added to the nuclear pellets, incubated on ice for 40 min, and centrifuged at 14,000 rpm for 10 min. Supernatants corresponding to the nuclear fraction were harvested. Protein concentrations of both cytoplasmic and nuclear fractions were determined, and all samples were analyzed by Western blot with 9% SDS-PAGE.
Anti-IFN-β neutralization
Medium containing either Nef (100 ng/mL) or IFN-β (200 UI/mL) were incubated for 1 h at 4°C with anti-IFN-β neutralizing antibodies (10−4 dilution of a preparation with a neutralizing titer of 4×106 U/mL). The medium was then thawed at 37°C for 30 min, and used to stimulate RAW264.7 cells. The cells were washed twice in PBS, collected, and lysed, and the total cellular lysates were assessed by Western blot for the tyrosine phosphorylation of both STAT1 and STAT2.
Results
recNef treatment induces STAT1 and STAT2 tyrosine phosphorylation in RAW264.7, but not in J774A.1 cells
We previously reported that the treatment of human monocyte-derived macrophages with recNef activates early intracellular signaling events (i.e., activation of NF-κB, MAPKs, and IRF-3), leading to the production of several cytokines and chemokines (i.e., interleukin-1β [IL-1β], IL-6, tumor necrosis factor-α [TNF-α], IFN-β, MIP-1α, and MIP-1β), starting after 2 h of treatment (13,25). These factors act in autocrine and paracrine manner, leading to tyrosine phosphorylation of STAT1, STAT2, and STAT3 (25 –27).
In an initial attempt to set up an in vitro murine monocyte/macrophage-based cellular system to study the effects of recNef treatment on these cellular populations, we chose two different monocytic cell lines: RAW264.7, which was isolated from an Abelson-derived tumor (28), and J774A.1, which was isolated from a murine histiocytic lymphoma (29). Both cell lines were isolated from BALB/C mice, are lysozyme-positive, and show phagocytic activity. The cells were treated for different times with wild-type recNef, and monitored for tyrosine phosphorylation levels of STAT1, STAT2, and STAT3. In the J774A.1 cell line, this treatment was unable to induce any significant increase in STAT1, STAT2, and STAT3 tyrosine phosphorylation levels, even at doses up to 500 ng/mL, and after 6 h of treatment (Fig. 1A and data not shown). Nevertheless, both murine recombinant IFN-β, and to a lesser extent LPS treatment of J774A.1, induced the rapid phosphorylation of STAT1, STAT2, and STAT3, ruling out the possibility that these cells are intrinsically unresponsive (Fig. 1B). Conversely, treatment of RAW264.7 cells with recNef induced the phosphorylation of both STAT1 and STAT2, that was clearly detectable after 2 h (Fig. 1C). Tyrosine phosphorylation of STAT1 and STAT2 persisted after 6/8 h of treatment. Dose-response analysis revealed that a concentration as low as 25 ng/mL of recNef is sufficient to induce tyrosine phosphorylation (Fig. 1D). Unlike human MDMs, recNef treatment was unable to induce any STAT3 tyrosine phosphorylation in RAW264.7 cells at all time points tested (Fig. 1E and data not shown). Nevertheless, recNef-treated RAW264.7 cells were able to release a STAT3-activating factor, as supernatants collected from these cells after recNef treatment were able to induce STAT3 tyrosine phosphorylation in J774A.1 cells (Fig. 1E). J774A.1 cells are sensitive to treatment with recombinant murine IL-6 (Fig. 1E), whereas RAW264.7 cells are not (data not shown), suggesting the possibility that the lack of STAT3 tyrosine phosphorylation following recNef treatment is due to the unresponsiveness of RAW264.7 to this cytokine. Therefore, we further characterized the response of the RAW264 cell line to recNef treatment.
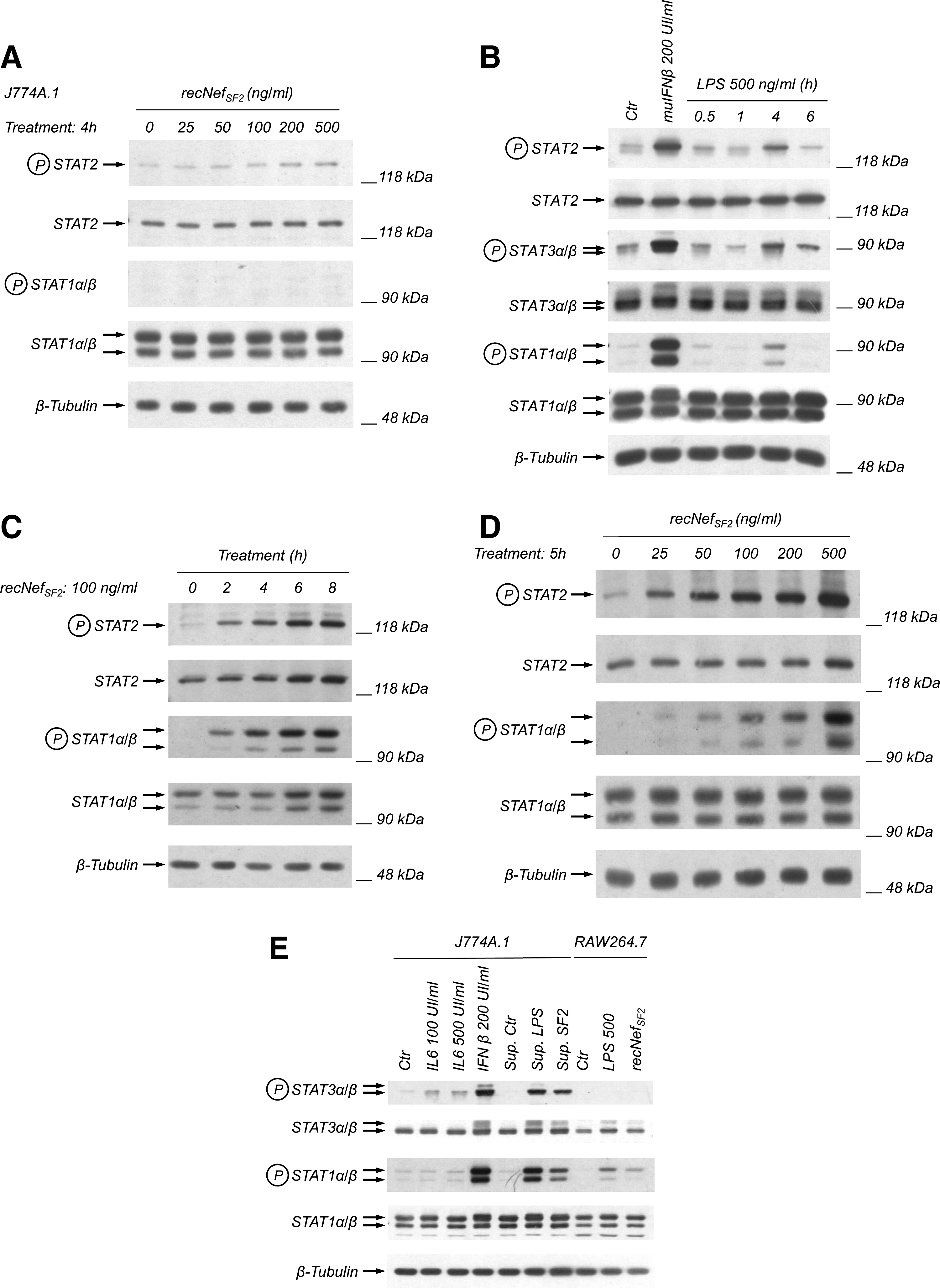
recNef treatment induces STAT1 and STAT2 tyrosine phosphorylation in RAW264.7 but not J774A.1 cells. (
Nef-induced NF-κB activation is essential for STAT1 and STAT2 tyrosine phosphorylation
In human MDMs, STAT1 and STAT2 tyrosine phosphorylation depends on Nef-dependent activation of both IKK-α and IKK-β (i.e., the two catalytic subunits of the Iκ-B kinase complex), and to a lesser extent, on the activation of p38 MAPK (25). To test if in murine macrophages STAT1 and STAT2 activation depends on these signaling pathways, we treated RAW264.7 cells with 100 ng/mL of wt recNef in the presence of specific kinase inhibitors. In particular we used BMS-345541, a specific IKK-activity inhibitor (33); the allosteric inhibitor of MEK-1/ERK PD98059 (30), the ATP binding competitive inhibitor of p38; SB203580 (31), and the ATP binding competitive inhibitor of JNK SP600125 (32). Pre-treatment of RAW264.7 with BMS-345541 completely abolished the activation of both STAT1 and STAT2 (Fig. 2). Pre-treatment of cells with both SB203580 and SP600125 decreased, but did not abolish, STAT tyrosine phosphorylation, whereas the ERK inhibitor PD98059 had no effect (Fig. 2). Taken together these results confirmed that IKK activity is essential for the production of STAT-activation factors, and that p38 and JNK play a role in fine tuning the activation of these factors, whereas ERK1/2 appears not to be involved in STAT activation.
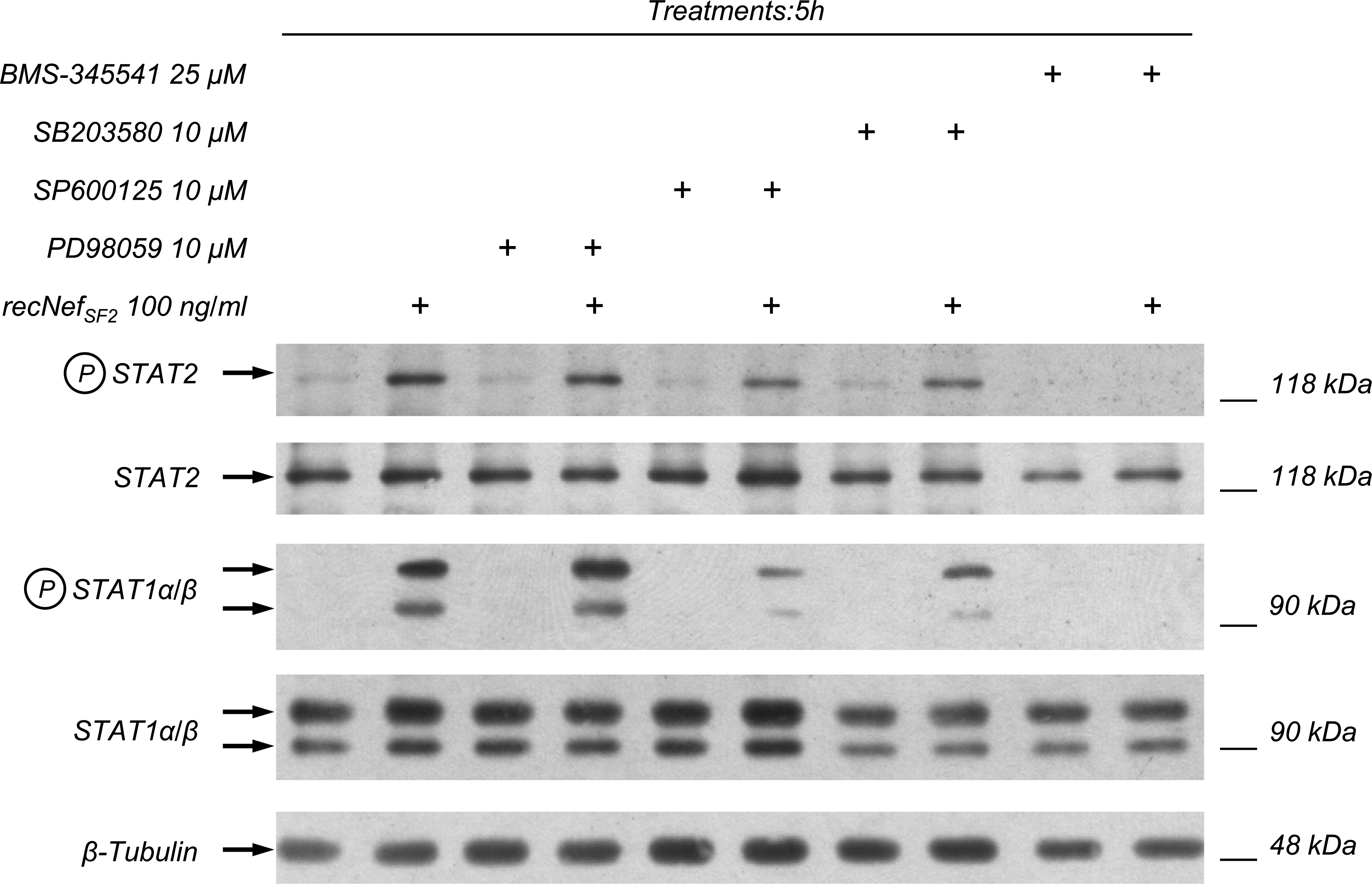
Nef-induced NF-κB activation is essential for STAT1 and STAT2 tyrosine phosphorylation. RAW264.7 cells were pre-treated for 1 h with the IKK inhibitor BMS-345541 (25 μM), the p38 MAPK inhibitor SB203580 (10 μM), the Jun N-terminal kinase inhibitor SP600125 (10 μM), or the MKK-1 inhibitor PD98059 (10 μM), and then incubated for 5 h with recNef (100 ng/mL). Total cellular extracts were analyzed by Western blot using anti-phospho-STAT1 and anti-phospho-STAT2. STAT1 and STAT2 expression were monitored using their respective antibodies, whereas the β-tubulin expression level was used as an internal loading control. One out of three independent experiments is shown.
recNef treatment induces JNK and p38 phosphorylation
As reported above, both SB203580 and SP600125 are able to decrease Nef-induced STAT1 and STAT2 tyrosine phosphorylation. These results suggest that p38 and JNK kinases are involved in the Nef-dependent upregulation of gene expression, leading to the synthesis and release of STAT-activating factors. To explore this possibility RAW264.7 cells were treated for different times with 100 ng/mL of wt recNef, and assessed by Western blot for the phosphorylation level of both MAP kinases. As shown in Fig. 3, recNef treatment induces the phosphorylation of both JNK and p38, although with different kinetics. JNK phosphorylation is indeed barely evident after 15 min of treatment, peaks at 30 min, and returns nearly to basal levels after 2 h (Fig. 3A). In contrast, p38 phosphorylation is clearly evident after 15 min, and was constantly augmented until 2 h (Fig. 3B). Again, these results parallel those obtained in human MDMs.
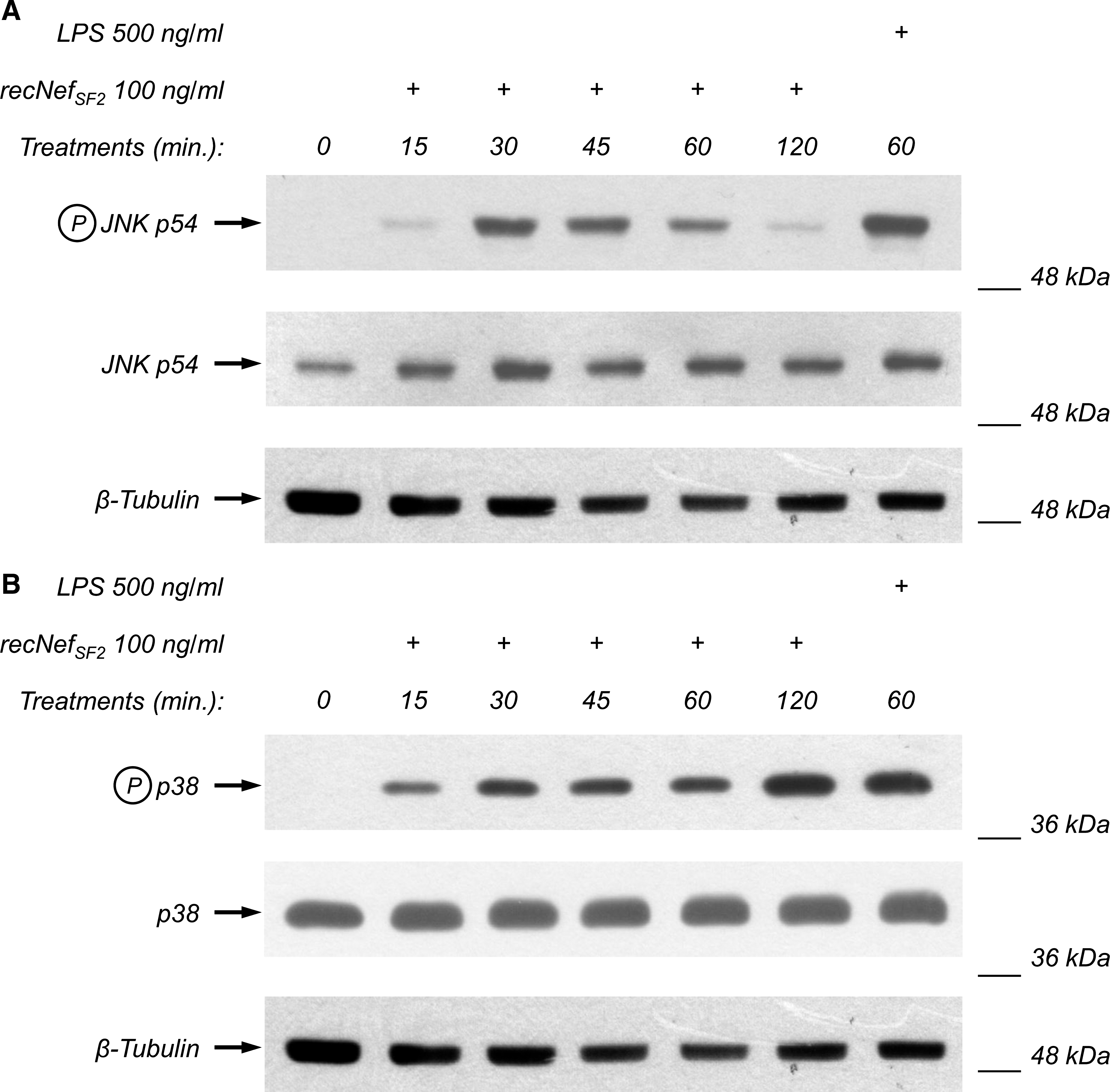
recNef treatment induces JNK and p38 phosphorylation. RAW264.7 cells were incubated for the indicated times with recNef (100 ng/mL) or, as positive control, for 60 min with LPS (500 ng/mL). Total cellular extracts were analyzed by Western blot using anti-phospho-JNK (
recNef treatment induces IKK-α and IKK-β phosphorylation, as well as Iκ-B-α degradation
The results reported in Fig. 2 and those obtained in human MDMs (25) demonstrate the fundamental role played by the NF-κB signaling pathway in the Nef-induced synthesis of STAT-activating factors. To evaluate the effect of recNef treatment of RAW264.7 on NF-κB signaling we stimulated the cells with 100 ng/mL of wt recNef for times ranging from 15 min to 2 h, and checked both the phosphorylation of IKK-α and IKK-β and the degradation of the inhibitor of NF-κB, namely I-κB-α. As a positive control cells were stimulated for 1 h with LPS at a concentration of 500 ng/mL. The results depicted in Fig. 4A show that recNef treatment of RAW264.7 cells induced rapid phosphorylation of both IKK-α and IKK-β which begins after 15 min and remains sustained for 2 h. Even if we were not able to evaluate the level of both IKK proteins due to the lack of antibodies that recognize in Western blot the murine form of these kinases, the appearance of a specific signal in response to LPS treatment was proof of IKK-α and IKK-β activation. The activation of IKKs was rapidly followed by the degradation of I-κB-α, which was evident at 30 and 45 min. At later time points (1 and 2 h), I-κB-α levels increased, because I-κB itself is a NF-κB-inducible gene (37). IKK activation and I-κB-α degradation were both directly correlated to the dose of recNef treatment used to stimulate the RAW264.7 cells (Fig. 4B).

recNef treatment induces IKK-α and IKK-β phosphorylation, as well as Iκ-B-α degradation. (
recNef treatment induces type I IFN-mediated events
Using an array procedure we have observed that treatment of human MDMs with recNef regulates the expression of many genes, and upregulates the level of IFN-β mRNA (13), a finding in agreement with the phosphorylation of IRF-3 (25), a constitutively expressed transcription factor, which is the main transcriptional regulator of the IFN-β gene. In addition, we reported that in human MDMs recNef treatment induced tyrosine phosphorylation of STAT2, an event occurring only in response to type I IFN-receptor engagement (25). As STAT2 tyrosine phosphorylation is also evident in RAW267.7 cells (Figs. 1C, 1D, and 2), we evaluated the possibility that recNef treatment of murine macrophages could also activate the IFN system. First, RAW264.7 cells were treated with 200 ng/mL of wt recNef, and IRF-3 phosphorylation was analyzed by evaluating the electrophoretic migration of the protein, with the slower-migrating form corresponding to the hyperphosphorylated one. Using total cytoplasmic extracts we detected only a slight modification of the migration pattern (data not shown). For this reason nuclear extracts of recNef-treated cells were analyzed by Western blot using anti-IRF3 antisera. The recNef treatment induced the appearance of two slight bands corresponding to the hyperphosphorylated forms of IRF-3 after 1 h (Fig. 5A, indicated by arrows). To further support these data, we treated RAW264.7 cells for 5 h with 100 ng/mL of recNef in the presence or absence of anti-IFN-β neutralizing antibodies, thereby checking the tyrosine phosphorylation of both STAT1 and STAT2. As a positive control, cells were stimulated with 200 IU/mL of IFN-β for 30 min in the presence or absence of anti-IFN-β neutralizing antibodies.
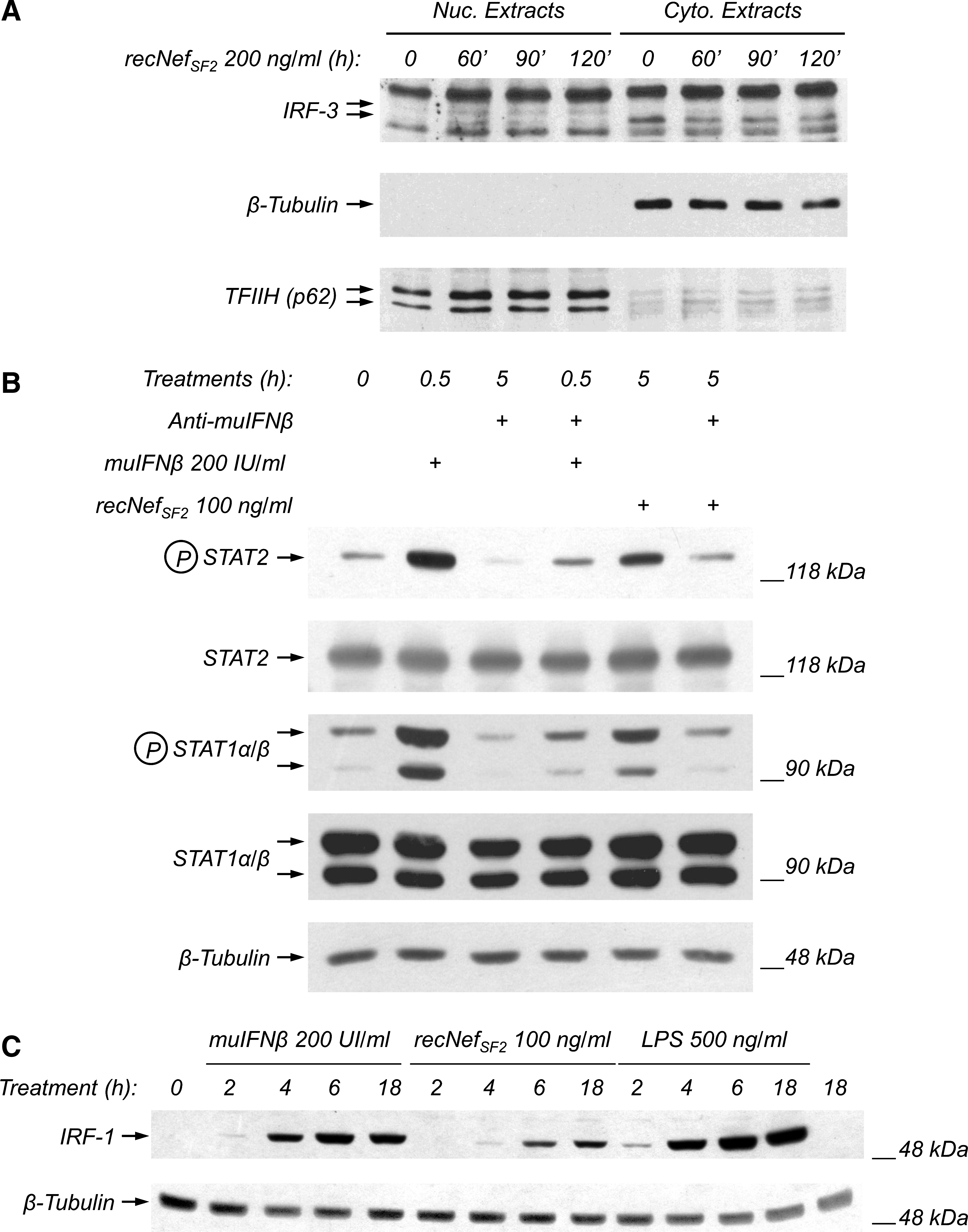
recNef treatment induces type I IFN-mediated events. (
As shown in Fig. 5B, anti-IFN-β neutralizing antibodies were able to completely prevent Nef-induced STAT1 and STAT2 tyrosine phosphorylation, indicating that both are completely due to the release of IFN-β. As further evidence of the Nef-dependent induction of type I IFNs, we analyzed if Nef treatment of RAW264.7 cells is able to upregulate the expression of IRF-1, an interferon-regulated gene that was initially identified as a transcriptional regulator of IFN-β-induced genes (38,39). As depicted in Fig. 5C, treatment of RAW264.7 with 100 ng/mL of wt recNef induces the appearance of a slight signal corresponding to IRF-1 after 4 h. This signal becomes apparent at 6 h, and is still evident after overnight treatment. As expected, both type I IFN (150 IU/mL), as well as LPS (500 ng/mL) treatment, upregulated the expression of IRF-1 (Fig. 5C). Collectively, these results indicate that the recNef treatment of RAW264.7 is able to induce the synthesis and release of IFN-β.
Myristoylation and acidic cluster are essential to achieve IKK and STAT activation
HIV-1 Nef exerts its pleiotropic effects in both T lymphocytes and monocyte/macrophages by interacting with several cellular partners using different sequence motifs (for review see 1, 40). In particular, it has been shown that Nef is targeted to the plasma membrane through co-translational myristoylation, and it is able to interact with SH3-bearing proteins through a poly-proline rich motif (PxxPxR). Instead, a CAWL motif is required for CD4 interaction, the N-terminal acidic cluster EEEE was reported to be involved in the Nef-dependent downregulation of HLA-A and HLA-B alleles (41), and the EE, LL, and DD amino acid sequences exposed in a C-terminal flexible loop of Nef (42) are required for the interaction with several elements of the endocytic machinery. To test if these sequences could play a role in the Nef-induced activation of NF-κB as well as of STAT1 and STAT2, we treated RAW264.7 cells with wt recNef or various mutants thereof (100 ng/mL each). In particular, we used a mutant in the myristoylation site (i.e., G2A), alanine-substituted CAWL, or PxxP, LLAA, DDAA, and EEAA mutants, and the 4EA mutant of the N-terminal acidic cluster EEEE. We also used a mutant with deletion of the first 44 amino acids, called ΔN-term, and a second mutant with deletion of the C-terminal loop, containing the three motifs involved in the interaction with the endocytic machinery, called ΔLoop (Fig. 6A). As reported, all mutants were able to induce both IKK phosphorylation (Fig. 6B), and STAT1 and STAT2 activation (Fig. 6C), with the exception of 4EA, the non-myristoylated G2A, and the ΔN-term ones. These results confirm previous observations in human MDMs (25,35), and underline the importance both of the myristoylation site and the N-terminal acidic cluster in the Nef-induced activation of the NF-κB and JAK-STAT pathways.
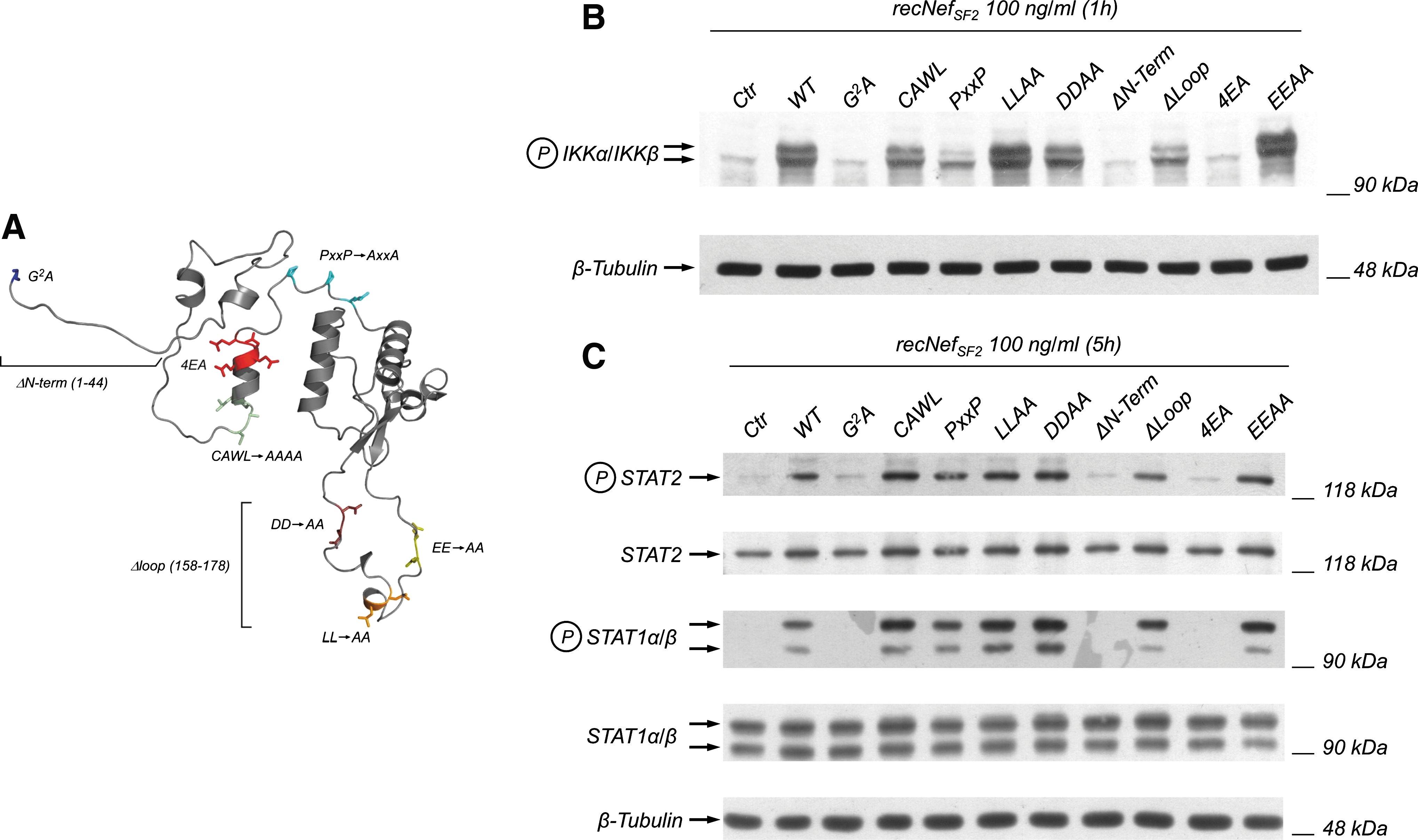
Myristoylation and an acidic cluster in Nef are essential to achieve IKK and STAT activation. (
recNef treatment induces STAT1, STAT2, and STAT3 tyrosine phosphorylation in primary macrophages
The herein reported results were obtained using the murine monocytic cell line RAW264.7, which was isolated from BALB/C mice (28). To assess if in primary murine monocyte/macrophages recNef treatment was able to induce the observed signaling effects, peritoneal macrophages were isolated from BALB/C mice and stimulated with wt recNef (Fig. 7). recNef treatment induced the tyrosine phosphorylation of STAT1, STAT2, and STAT3 after 2 h at both 100 ng/mL and 500 ng/mL (Fig. 7). STAT activation is still present after 6 h, and is consistent with the phosphorylation detected in these cells after a 6-h treatment with LPS (Fig. 7). These results confirm those obtained for the macrophage cell line RAW264.7, and also demonstrates that in primary macrophages STAT3 can be tyrosine phosphorylated after recNef treatment.

recNef treatment induces STAT1, STAT2, and STAT3 tyrosine phosphorylation in primary macrophages. Purified primary peritoneal macrophages were incubated for the indicated times with recNef (100 ng/mL) or, as positive control, with LPS (500 ng/mL) or muIFNβ (200 IU/mL for 30 min). Total cellular extracts were analyzed by Western blot using anti-phospho-STAT1, anti-phospho-STAT2, and anti-phospho-STAT3 antibodies. The β-tubulin expression level was used as an internal loading control. Results shown here represent one out of two independent experiments.
Discussion
Increasing evidence demonstrates the capacity of Nef to modulate several cellular processes by interacting with cellular counterparts. This property allows Nef not only to improve the survival of infected cells by interacting with cellular intermediates of the apoptotic signaling pathways (43), but also to modulate the effector functions in infected T lymphocytes and macrophages. In particular, in the latter cellular population, we and others have shown that both exogenously-added or endogenously-expressed Nef is able to induce the synthesis and release of several proinflammatory cytokines and chemokines (12,13,44), probably with the ultimate goal of recruiting target cells and enhancing the spread of infection. From this point of view it has been recently reported that Nef is able to promote the infection of resting T lymphocytes by acting on NF-κB pathways in macrophages, thereby inducing the release of sICAM1 and sCD23 from these cells (14). Similarly, in immature dendritic cells it has been reported that exogenous- or endogenously-expressed Nef is able to induce the synthesis and release of IL-6, IL-12, TNF-α, MIP-1α/CCL3, MIP-1β/CCL4, and CXCL8/IL-8 (45,46), whereas only exogenous Nef induces maturation of dendritic cells (45,47). Overall, these data underline that the proinflammatory Nef function in the myeloid compartment acts regardless of its source (i.e., exogenously-added versus endogenously-expressed), a striking difference with the lymphoid compartment. On T cells, endogenously-expressed Nef plays a positive role by interacting with TCR signaling elements, thereby lowering the activation threshold in infected CD4+ lymphocytes (8,48), whereas exogenous added Nef is essentially a “death signal” for CD4+ T cells, and induces apoptosis through interactions with CXCR4 (49,50).
The study of Nef's effects on signaling in monocyte/macrophages presents many technical limitations. Overexpression approaches using plasmids encoding dominant negative, dominant positive, or kinase dead proteins cannot easily be done in monocyte/macrophages, as they are difficult to transfect. siRNA approaches, allowing the silencing of specific target proteins, may be applied only in monocytic cell lines, as they are not efficient in primary MDMs, which are a resting cell population. In addition, viral expression vectors may induce specific responses such as, for example, the production of type I IFN, that in turn are able to activate signaling pathways, which may interfere with the interpretation of the results. One way to obviate these problems is via the use of monocyte/macrophages derived from knock-out or knock-in mice. It is noteworthy that Hanna and colleagues showed that Nef transgenic mice develop an immunodeficiency syndrome, which closely resembles AIDS, demonstrating that at least in this transgenic system, Nef is active and appears to be a major determinant of pathogenicity (3,51). Nevertheless, the effects of Nef expression on macrophages derived from transgenic mice, as well as the pattern of proinflammatory factors produced by these cells in Nef-transgenic mice, were only slightly characterized. For these reasons we tried to set up a murine macrophage-based cellular system that would allow us to study in more detail the effects previously described in human MDMs (25 –27,35). Indeed, we have previously shown that Nef treatment of MDMs replicates some of the effects induced by Nef during HIV-1 infection. To characterize the effects of Nef treatment on murine monocyte/macrophage signaling pathways, we initially utilized two different cell lines, RAW264.7 and J774A.1, to examine STAT1, STAT2, and STAT3 tyrosine phosphorylation as a marker of response. In fact, this effect is induced in human MDMs as a consequence of the rapid induction of a set of proinflammatory cytokines and chemokines that act in autocrine and paracrine manner. As shown in Fig. 1, only RAW264.7 responded to recNef treatment in terms of STAT1- and STAT2-induced tyrosine phosphorylation, whereas J774A.1 cells were unresponsive. Control experiments performed to evaluate the tyrosine phosphorylation of STAT1, STAT2, and STAT3 following muIFN-β and LPS treatments demonstrated that these signaling intermediates could be activated in J774A.1, both directly (i.e., by IFN-β treatment), and through the synthesis and release of activating factors (i.e., by LPS treatment). The reasons for the differing behavior of these two cell lines is unknown. As both cell lines were isolated from BALB/C mice, we can rule out that these differences are due to the genetic background of the mice from which they were derived. However, we cannot exclude the possibility that the different characteristics of these cell lines in terms of their capacity to incorporate recNef and/or effector functions may explain the lack of STAT activation in J774A.1 cells following recNef treatment.
Another striking difference between human macrophages and the murine cell line RAW264.7 is the lack of STAT3 tyrosine phosphorylation after Nef treatment. Again, muIFN-β treatment of RAW264.7 cells induces STAT3 tyrosine phosphorylation (data not shown), indicating that STAT3 can be activated. Control experiments performed by treating both RAW264.7 and J774A.1 with recombinant muIL-6 or with conditioned supernatants collected from recNef-treated RAW264.7 cells (Fig. 1B), as well as those obtained in recNef-treated peritoneal primary macrophages (Fig. 7), indicate that the RAW264.7 cell line is unable to respond to IL-6. One of the main STAT3-activating factors among the several cytokines and chemokines produced by human MDMs after recNef treatment is IL-6, and according to the results obtained in MDMs, recNef treatment is also able to induce STAT3 phosphorylation in primary murine macrophages (Fig. 7).
Regarding the early signaling events triggered by recNef after 15 to 60 min of treatment of primary human MDMs (i.e., MAPKs, IKKs, and I-κB degradation), they were all confirmed in RAW264.7 cells (Figs. 3 and 4), demonstrating that there is no difference between human and murine macrophages in terms of Nef-induced early signaling features, with the exception of IRF-3, the main transcriptional regulator of IFN-β gene expression. Indeed, IRF-3 activation was detected only at very low levels in RAW264.7 cells, and was delayed compared to the results seen in human MDMs (Fig. 5A). This observation is in agreement with the difficulty to detect antiviral activity in supernatants collected from Nef-treated RAW264.7 cells using a biological assay measuring resistance to EMCV infection (data not shown). Nevertheless, STAT2 tyrosine phosphorylation, a specific hallmark of the type I IFN response, was achieved in recNef-treated RAW264.7 cells (Figs. 1 and 2), and was prevented by using anti-IFN-β antiserum, indicating that type I IFN is produced, although at very low levels (Fig. 5B). Neutralization experiments not only confirmed that the synthesis and release of type I IFN is responsible for Nef-induced STAT2 tyrosine phosphorylation, but also that the type I IFN produced in these cells is IFN-β, similar to previous results we obtained in human MDMs and the monocytic cell line THP-1 (25,35). Using a series of Nef mutant proteins, here we confirmed results previously obtained in human MDMs, which underline the importance of both the myristoylation and the N-terminal acidic cluster of Nef in the signaling activity induced by the viral protein. Interestingly, we have recently reported that TRAF2 and TRAF6 silencing abolished the Nef-induced tyrosine phosphorylation of STAT1 and STAT2 in THP-1 monocytic cells. In addition, Nef is able to interact in vitro with TRAF2, but not TRAF6, and the acidic cluster is also involved in this interaction (35). These results suggest that Nef interferes with the cell receptor signaling that regulates the expression of proinflammatory cytokines and chemokines and IFN-β, such as CD40 and TLR, which act on specific TRAFs, and result in the activation signals observed.
Another similarity between human and murine systems is the kinetics of the induction of signaling cascades (see model in Fig. 8). Exogenously-added Nef is indeed able to activate a set of “early signals” within few minutes (i.e., 15 to 45 min), based on the activation of NF-κB, p38, and JNK kinases. These events lead to the synthesis and release of several proinflammatory factors and IFN-β, leading to “late signals” (i.e., from 1 h 45 min to 2–8 h or more), based on STAT tyrosine phosphorylation. Data obtained after prolonged incubation with Nef (24–48 h) indicate the presence of succeeding waves of STAT-based signals (G. Mangino, unpublished data), suggesting the establishment of a Nef-dependent persistent proinflammatory state in myeloid cells. The results reported here show that the murine system appears to be a suitable model system to further investigate the molecular mechanisms of the action of Nef, and that both RAW264.7 and primary peritoneal macrophages can be used in studies performed in human and/or simian models.
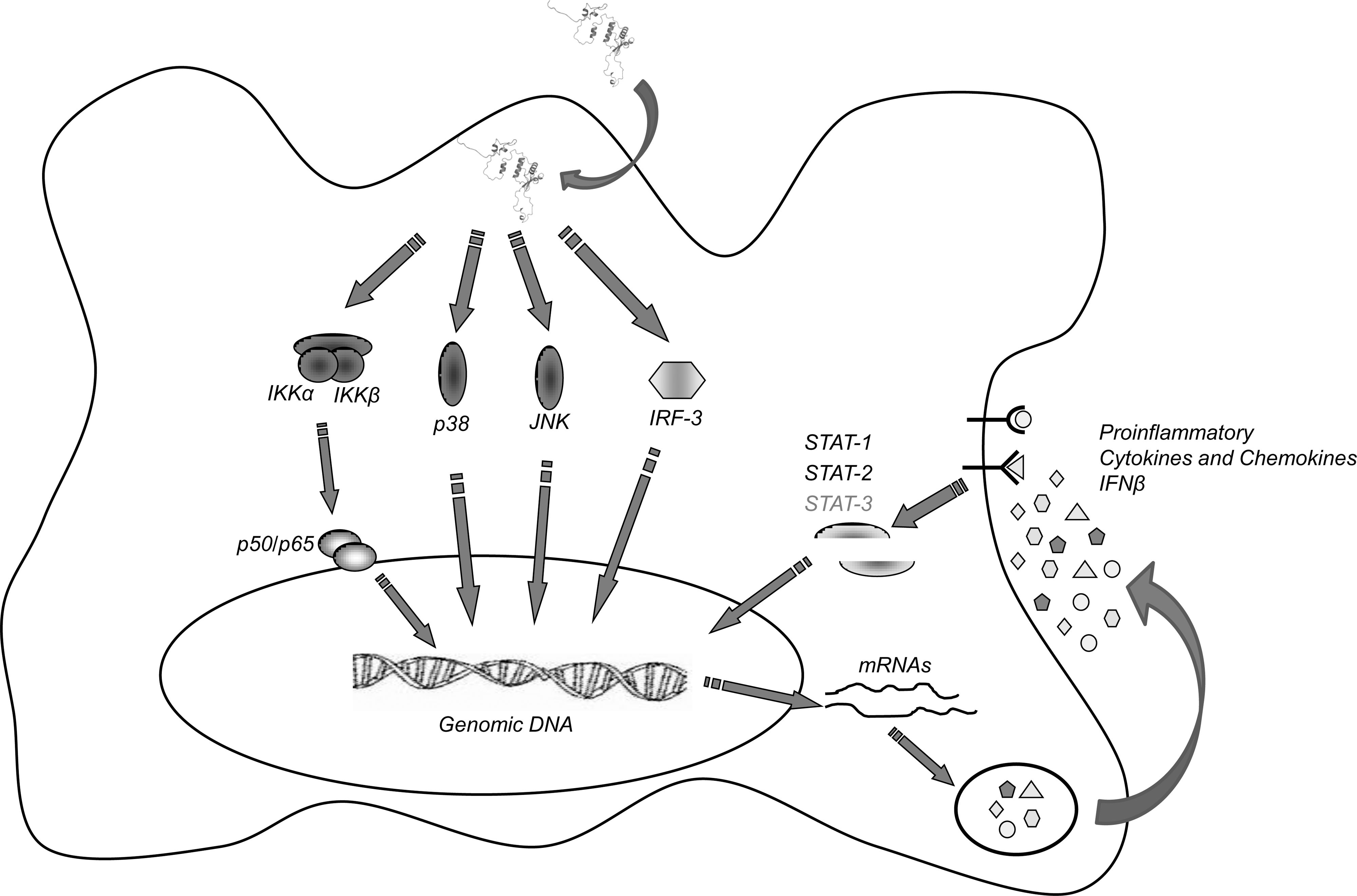
Model for Nef-induced signaling in murine macrophages. Exogenous Nef is internalized and induces the rapid activation of IKK/NF-κB, p38, JNK, and IRF-3. These signals cause a first wave of cellular gene activation that results in the synthesis and release of proinflammatory factors and in the production of IFN-β. After binding their specific receptors on the same or neighboring cells, these factors lead to the activation of STAT1, STAT2, and STAT3, further regulating cellular functions. STAT3 is shown in gray, as it is tyrosine phosphorylated in primary macrophages, but not in RAW264.7 cells (modified from 25).
Footnotes
Acknowledgments
This work was supported by grants from the Italian National Research Program on AIDS of the Istituto Superiore di Sanita’ (Affabris N.40.G1), Rome, Italy, and from PRIN-MIUR (Progetti Rilevante Interesse Nazionale–Ministero dell'Universita’ e della Ricerca Scientifica) 2007 (Affabris prot.20072J9RWM_002), Italy.
Author Disclosure Statement
No competing financial interests exist.