
Abstract
Swine influenza virus (SIV) is an important viral pathogen in pig populations. However, commercial vaccines cannot provide complete protection with induced humoral immunity only, and require frequent updates to fight against current isolates. DNA vaccination is an effective means of eliciting both arms of the immune system, the humoral and cellular immune responses. In this study, DNA vector pcDNA3.1 was inserted with a chimeric intron downstream of the CMV promoter region followed by a Kozak sequence to enhance the expression of gene inserts. The C-terminal of the VP22 gene (VP22c), encoding the tegument protein of bovine herpesvirus-1, was fused separately to the N-terminal of four quadruplicated epitopes: two B-cell epitopes (HA91-108 and M2e), and two T-cell epitopes (NP366-374 and NP380-393), which were conserved, at least among the three SIV subtypes prevailing in pig populations in North America. Linker -KK- was used to space between each copy of the two B-cell epitopes, and -RVKR- was used for the two T-cell epitopes, in order to enhance the presentation of epitopes to the immune system. The expression of epitopes was confirmed in in vitro transfection of 293FT cells, and higher percentages of epitope-positive cells were achieved from the plasmids containing VP22c than those without. After the DNA plasmids were administered to mice intramuscularly in combination or separately, or boosted with recombinant proteins of quadruplicated epitopes fused to VP22c, the vaccine stimulated the desired epitope-specific humoral immunity to the two B-cell epitopes, and cellular immunity to the epitope NP380-393. Our results indicate that plasmids with quadruplicated epitopes fused to the VP22c may be a potential vehicle in developing epitopes as vaccines against SIV.
Introduction
Current swine influenza vaccines in North America are inactivated whole-virus vaccines. They can induce only strain-specific humoral immune responses, while broad-spectrum humoral and cellular immunity are essential for providing cross-protection against heterologous viruses, which is highly desirable for fast-evolving influenza viruses (5,6). DNA vaccines have a great potential to achieve this purpose (7). DNA vaccines can induce both humoral and cellular immunity to gene inserts in DNA vectors because they can be easily modified in a variety of ways to achieve different purposes. To achieve a broad-spectrum immunity, epitope vaccines have been explored in order to develop a universal influenza vaccine. With epitopes conserved among multiple subtypes of influenza virus, it may be able to achieve the desired broad protection. However, epitopes are too short to be immunogenic without an appropriate carrier or adjuvant. Inserting these epitopes into DNA vectors may be a potential way to reach the goal.
To develop an effective DNA vaccine against SIVs, multiple approaches were applied in our study to optimize the immunogenicity of DNA vaccines: (1) selecting four consensus immunogenic epitopes conservative to the major subtypes of SIVs (H1N1, H1N2, and H3N2), including two T-cell (NP366-374 and NP380-393), and two B-cell epitopes (HA91-108 and M2e) (H3 numbering) (8,9); NP366-374, NP380-393, and M2e are almost 100% conservative in all SIVs through BLAST in NCBI; HA91-108 is nearly 100% conservative in H1-subtyped SIVs, while the several amino acids needed for receptor binding are conservative in H3 SIVs (10); (2) enhancing antigen expression by applying codon optimization (11), and insertion of the Kozak sequence (12,13) and a chimeric intron (14,15); (3) increasing the number of target protein-presenting cells by fusing the epitopes with the C-terminal portion of the VP22 gene (VP22c) from bovine herpesvirus-1 to enhance intercellular migration of fused proteins (16); (4) augmenting antigen presentation by quadruplicating epitopes (17 –19), linked with linker sequences targeting peptide presentation by MHC class I (20 –22), or MHC class II molecules (23 –25); and (5) applying the heterologous DNA prime-protein boost regimen to enhance immune responses (26).
As measured by both immunofluorescence assay and flow cytometry, in vitro experiments confirmed the expression of genes from plasmids, and higher numbers of epitope-positive cells were achieved from those plasmids with fusion to the C-terminal of VP22c than those without. Plasmids containing VP22c-eptiope chimera were applied intramuscularly and purified recombinant proteins were applied subcutaneously to mice. The peptide-specific humoral immunity was assayed by ELISA, and cellular immunity indicated by IFN-γ secretion was measured by flow cytometry. In this study, the vaccine strategy stimulated the desired immunity in mice, and can serve as a platform for generating DNA-epitope vaccines for influenza viruses.
Materials and Methods
Mice
BALB/c mice at the age of 6–8 wk were purchased from Harlan Sprague Dawley (Indianapolis, IN) and used for vaccination study. All mice were maintained with free access to sterile food and water. The experimental protocol was approved by the Purdue Animal Care and Use Committee.
Construction of DNA plasmids with quadruplicated epitopes
Restriction enzymes and T4 DNA ligase were all from New England Biolabs (Ipswich, MA). DNA vector pcDNA3.1/V5-His (Invitrogen, Carlsbad, CA) was inserted at the Hind III restriction enzyme site with a chimeric intron sub-cloned from vector pCI (Promega, Madison, WI). The resulting vector was named pIA. Viral DNA from bovine herpesvirus-1 was extracted with the QIAamp DNA minikit (Qiagen, Valencia, CA) from cultured virus. VP22c gene (amino acids 121–258) was amplified by PCR from bovine hepesvirus-1 with High Fidelity Platinum® Taq DNA Polymerase (Invitrogen), and flanked by restriction enzymes Kpn I and Xho I. The primer sequences were VP22c-F:
In vitro transfection of DNA plasmids on 293FT cells
The 293FT cells (Invitrogen) were cultured in DMEM (Invitrogen), supplemented with 10% FBS (Atlanta Biologicals, Lawrenceville, GA), 1×non-essential amino acids (Invitrogen), 1×sodium pyruvate (Invitrogen), 1×GlutaMAX (Invitrogen), and 1% HEPES (Invitrogen). DNA plasmids mixed with lipofectamine 2000 (Invitrogen) at an N/P of 5 were added to 293FT cells in 2 mL cell suspension at a density of 1×106 cells per well in 6-well cell culture plates coated with poly-D-lysine (Becton Dickinson Labware, Bedford, MA). The plates were examined after 48 h incubation in a 37°C incubator with 5% CO2.
Immunofluorescence assay (IFA)
One microgram of DNA plasmids was transfected into 293FT cells per well in 6-well cell culture plates for IFA. Cells were fixed with 80% acetone for 5 min, and rinsed with PBS (pH 7.4). Anti-His (C-term) monoclonal antibody (Invitrogen) at a dilution of 1:200 was applied to the fixed cells, followed by fluorescein labeled goat anti-mouse IgG (H+L; KPL, Gaithersburg, MD) at a dilution of 1:60. The staining was observed with a Nikon Eclipse TE2000-S (Nikon Instruments, Melville, NY).
Western blotting
Four micrograms of DNA plasmids were transfected into 293FT cells per well in 6-well cell culture plates for Western blot analysis. Cells were lysed by sonication and lysate was centrifuged at 10,000 g for 10 min. After being mixed with Tricine Sample Buffer (Bio-Rad Laboratories, Hercules, CA) and boiled for 5 min, the supernatant was loaded on pre-cast PAGEr® Gold Gels (Lonza Rockland, Rockland, ME), and transferred to nitrocellulose membranes (Invitrogen). After being blocked with 5% milk in PBST (PBS with 0.1% Tween 20), the protein was stained with Anti-His (C-term) monoclonal antibody (Invitrogen) at a dilution of 1:2000, and peroxidase-labeled goat anti-mouse IgG (H+L; KPL) at a dilution of 1:2000. TMB-1 Component Membrane Peroxidase Substrate (KPL) was used to develop the stained membrane.
Flow cytometry
Four micrograms of DNA plasmids were transfected into 293FT cells for flow cytometric analysis. Cells were harvested 48 h after transfection and treated with Accutase (Sigma-Aldrich, St. Louis, MO) to yield a single-cell suspension. Cells were fixed and permeabilized with BD Cytofix/Cytoperm Buffer (BD Biosciences, San Jose, CA), followed by staining with Anti-His (C-term) monoclonal antibody at a dilution of 1:100, and fluorescein-labeled goat anti-mouse IgG (H+L) at a dilution of 1:100. Stained cells were analyzed with a BD FACSCanto II flow cytometer and FACSDiva™ v 6.1.2 software (BD Biosciences).
Production of recombinant quadruplicated epitopes
The fused genes of VP22c-epitope chimera were sub-cloned into vector pET30a+ (Novagen, Madison, WI) for expression of recombinant proteins. The resulting plasmids were transformed into Escherichia coli strain BL21-CodonPlus (Stratagene, La Jolla, CA). When OD 600 of bacteria culture reached 0.4–0.6, an optimized concentration of IPTG at 0.1 mM was added, and bacteria were cultured for another 4 h at 30°C. The pelleted bacteria were resuspended in BugBuster Protein Extraction Reagent, and supplemented with 10 μL Lysonase Bioprocessing Reagent (Novagen) per gram of cell paste. After being centrifuged at 16,000 g for 20 min, the supernatant was transferred to His-Bind Columns following the manufacturer's instructions for His-Bind Kits (Novagen). After being dialyzed in Spectra/Por CE Float-A-Lyzer G2 tubes (Spectrum Laboratories, Rancho Dominquez, CA) against PBS buffer for 24 h, the eluted protein was concentrated with Amicon Ultra-15 centrifugal filter devices (Millipore, Billerica, MA) by centrifuging at 2000 g for 10 min. The protein concentration was measured with a Quick Start Bradford Protein Assay Kit (Bio-Rad Laboratories). Purified protein was confirmed by Western blotting.
Immunization procedures
The mouse study design is summarized in Table 1. There were 20 mice in group 1 (G1), and 10 each in groups 2–7 (G2–G7). They were vaccinated two or three times in the tibialis anterior muscles with DNA vaccine, and subcutaneously in the back of the neck with recombinant protein, in 100-μL volumes 3 wk apart. The mice in G1 were inoculated with 100 μL PBS per injection. Mice were vaccinated twice in G3 or three times in G2 with the combination of the four plasmids pVP22cHA91, pVP22cM2e, pVP22cNP366, and pVP22cNP380 at 25 μg each. The third dose in G3 was 12 μg of a mixture of recombinant proteins (3 μg per protein) mixed with 12 μg of ISCOM AbISCO-100 (ISCONOVA AB, Uppsala, Sweden). Serum from mice in G2 and G3 was collected from the submandibular veins at study day 42 (D42) and day 60 (D60). Mice in G4–G7 were vaccinated with different plasmids in two doses at study day 0 (D0) and day 21 (D21). Serum was collected at D42 and tested positive for epitope-specific antibody response by ELISA, as discussed below. The mice in G4–G7 and half of the mice in G1 were euthanized with CO2 at D42 for measurement of the cellular immunity by flow cytometry. All the remaining mice were euthanized at D60 and cellular immunity was also measured in these mice.
Intracellular cytokine staining assay (ICCS) for IFN-γ
IFN-γ-secreting CD8+ T cells were detected as an indicator of cellular immunity. Spleens harvested from mice euthanized with CO2 were placed in a 70-μm cell strainer (BD Biosciences) and dissociated with a plunger from a 5-mL syringe. Splenocytes were then incubated in 3 mL ACK lysing buffer (BD Biosciences) for 5 min at room temperature. After being washed with RPMI 1640 (Invitrogen), supplemented with 10% FBS (Atlanta Biologicals), 1×penicillin-streptomycin-glutamine (Invitrogen), 1×non-essential amino acids (Invitrogen), and 1×sodium pyruvate (Invitrogen), the splenocytes were re-suspended in this medium at a concentration of 1×106 cells/mL, seeded into 12-well plates in 2 mL per well, and cultured in a 37°C incubator with 5% CO2. Splenocytes were stimulated with 10 μg of synthesized peptides (Genscript) per milliliter of medium for 6 h with the presence of 2 μL/mL of GolgiStop protein transport inhibitor (BD Biosciences) for the last 4 h of incubation. Stimulated splenocytes were harvested, blocked with Fc Block (BD Biosciences), and stained with PerCP/Cy5.5 anti-mouse CD4 antibody, and FITC anti-mouse CD8a antibody (BioLegend, San Diego, CA), followed with treatment by Cytofix/Cytoperm Buffer (BD Biosciences), and staining with APC anti-mouse IFN-γ antibody (BioLegend). Data were then acquired with a BD FACSCanto II flow cytometer (BD Biosciences). The lymphocyte population was gated in the scatterplots of the whole cell population. The percentage of IFN-γ+ lymphocytes were shown as the percentage of CD4+ or CD8+ cell populations.
Epitope-specific enzyme-linked immunosorbent assay (ELISA)
High-protein binding 96-well microplates were coated with 10 μg of synthesized epitopes (GenScript) diluted in 100 μL carbonate-bicarbonate buffer (pH 9.6), in columns with odd-numbered numbers, and coating buffer only in even-numbered columns, at 4°C overnight. After blocking with Block Ace (AbD Serotec, Raleigh, NC) for 1 h at room temperature, 100 μL serum at 1:50 dilution in Block Ace was applied to each well and incubated at 37°C for 1 h. For each serum sample, it was applied to both the coated and uncoated wells side by side in a plate. After washing with PBST buffer three times, peroxidase-labeled goat anti-mouse IgG monoclonal antibody (KPL) was applied to each well at a dilution of 1:2000 with Block Ace and incubated at 37°C for 1 h. Following washing three times with PBST, 100 μL of SureBlue Reserve TMB substrate (KPL) was added to each well, and 100 μL of Stop solution (KPL) was applied after 5 min of incubation at room temperature. The plates were read at optical density (OD) 450 with an EMax™ microplate reader (Molecular Devices, Sunnyvale, CA). The OD 450 values for each sample were calculated as the value from the coated well subtracted by that from the uncoated well. The epitope-specific antibody levels were reported as the mean OD 450 of each group. Samples were considered positive when the mean ODs were higher than 0.200.
Microneutralization (MN) assay
MDCK cells were cultured in DMEM (Invitrogen), supplemented with 10% FBS (Atlanta Biologicals), 1×non-essential amino acids (Invitrogen), 1×sodium pyruvate (Invitrogen), 1×GlutaMAX (Invitrogen), and 1×HEPES buffer (Invitrogen). MDCK cells were seeded in 96-well cell culture plates at a density of 5×104 cells per well. On the next day, twofold serial dilutions (starting from 1:10) of serum in duplicate were made in 50 μL of SIV rinsing medium (DMEM supplemented with TPCK-trypsin; Sigma-Aldrich, at a final concentration of 2 μg/mL). The diluted serum samples were mixed with 100 TCID50 of SIV (A/swine/Indiana/67/2007, H1N1) in 50 μL of SIV rinsing medium, and incubated at 37°C for 1 h. The cells were rinsed three times with SIV rinsing medium. The mixture of serum-virus was added into each well and incubated at 37°C for 1 h. After the incubation, the cells were washed three times with SIV culturing medium (DMEM supplemented with 1×non-essential amino acids, 1×sodium pyruvate, 1×GlutaMAX, 1×HEPES buffer, TPCK-trypsin at a final concentration of 2 μg/mL, and BSA trypsin [Sigma-Aldrich] at a final concentration of 0.2%). The cells were incubated in 100 μL of SIV-culturing medium per well at 37°C for 3 d. MN titer was defined to be the highest dilution that completely inhibited virus infection.
Statistical analysis
One-way ANOVA followed by pair-wise comparison with Tukey's test was applied to the data analysis. A p value of<0.05 was considered significant.
Results
Confirmation of the expression of DNA plasmids in 293FT cells
DNA constructs are schematically shown in Fig. 1A. To confirm the expression of fused genes, plasmids were transfected into 293FT cells. Several assays were utilized to detect the expression of fused genes via monoclonal antibody against 6XHis tag. The expression of fused genes was first confirmed by immunofluorescence assay as shown in Fig. 1B. In order to see the intercellular trafficking effect of VP22c fused to the selected epitopes, 1 μg of plasmids were transfected onto 293FT cells in 6-well cell culture plates. There were only sparsely distributed positive cells in those wells that were transfected with plasmids without VP22c (Fig. 1B c, e, g, and i), compared to the corresponding genes with it (Fig. 1Bd, f, h, and j). So VP22c was capable of transporting genes that were fused with it to adjacent cells. To get numerical data about the transfection with different plasmids, flow cytometry was applied to measure the percentage of epitope-positive cells 48 h after transfection with 4 μg of plasmids per well in 6-well cell culture plates. As shown in Fig. 1C, the dot plots were acquired from flow cytometry and average percentages of epitope-positive cells are shown in each plot representative of three repeated experiments. The data from Fig. 1C are summarized in Fig. 1D, and statistical analyses were conducted on the data. A significantly higher (p<0.001) percentage of epitope-positive cells was achieved from those plasmids with VP22c, compared to those without it. Therefore, it was consistently confirmed that VP22c-fused epitopes migrated from transfected cells into surrounding cells. Western blotting was also applied to confirm the expression of fused genes. As shown in Fig. 2A, the fused protein products with the desired molecular weights were detected in those cells transfected with plasmids containing VP22c. However, we did not detect the desired protein from cells transfected with plasmids that did not contain VP22c, because of the very low percentage of epitope-positive cells, as shown in Fig. 1C. With consistent data acquired from the three assays, the plasmids with VP22c were selected for the following experiments.
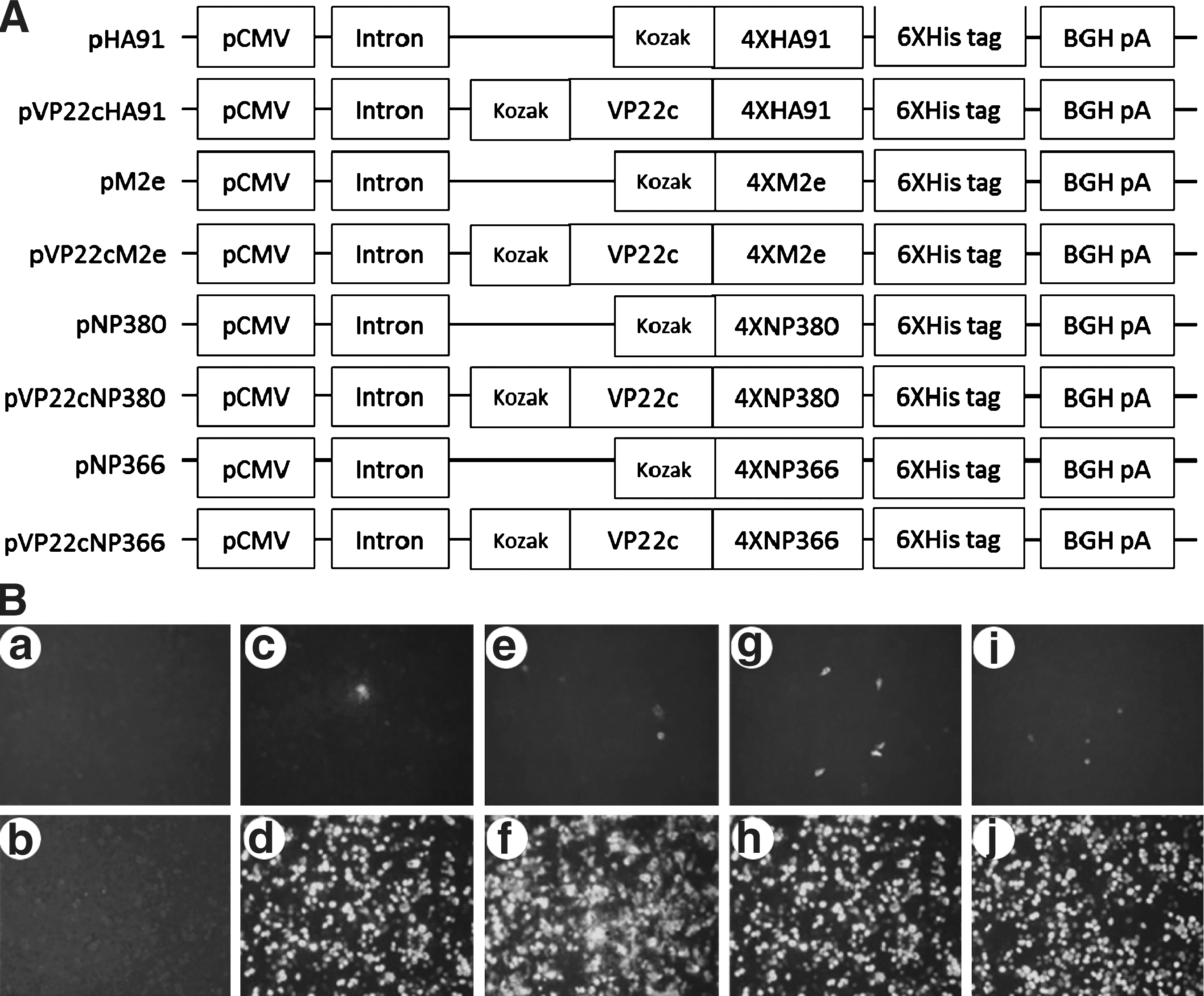
Schematic diagram of DNA plasmids and the in vitro conformation of protein expression. (

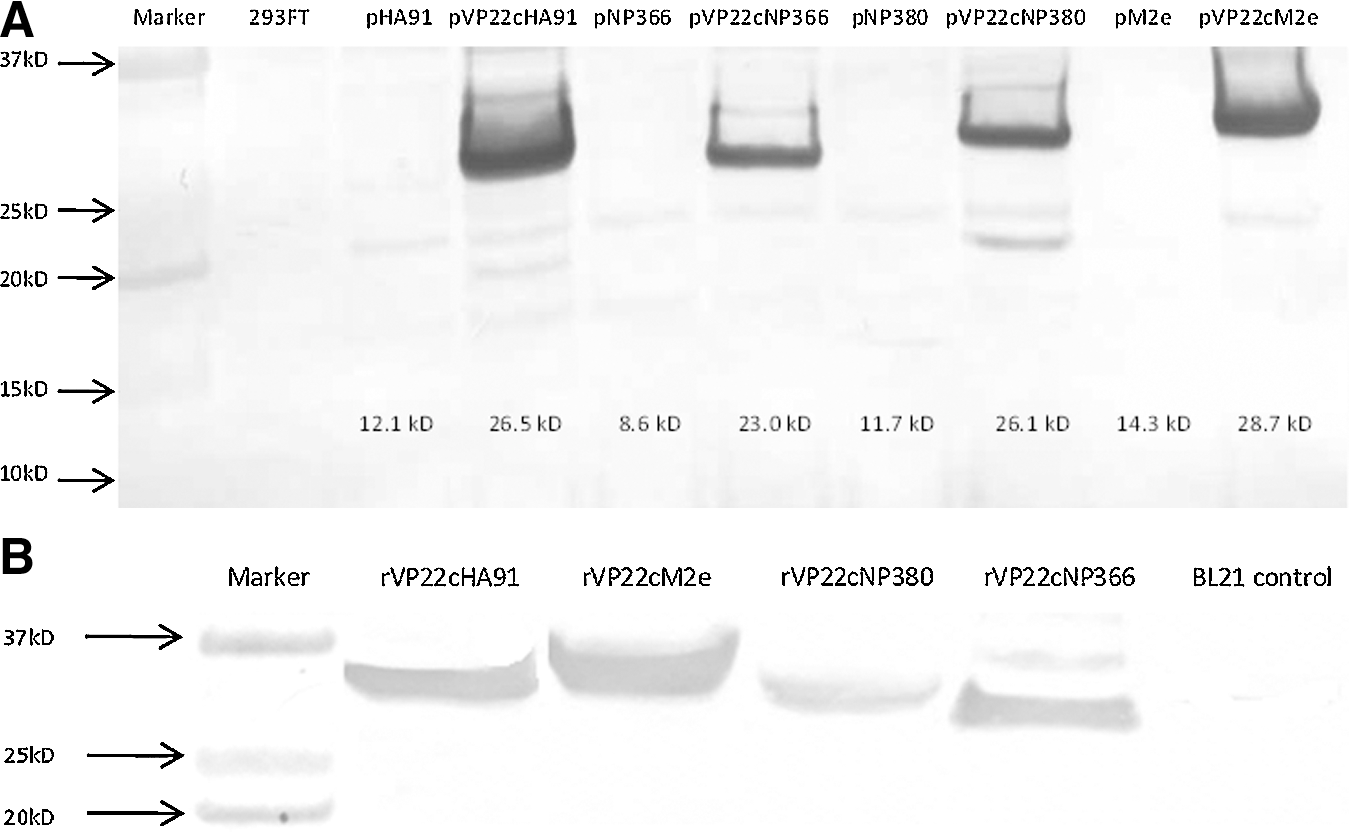
Western blotting confirmed the expression of inserts from plasmid-transfected 293FT cells in
Production of recombinant quadruplicated epitopes
The VP22c-epitope fused genes were sub-cloned into expressing vector pET30a+ and expressed in BL21-CodonPlus bacteria. After the expressing condition was optimized, the fused genes were expressed in a larger volume. The purified protein was detected by Western blotting as shown in Fig. 2B.
Intracellular cytokine staining assay for IFN-γ
The cellular immune response upon vaccination was indicated by detecting the IFN-γ-expressing cells with flow cytometry. The mice from G4–G7 and half of the mice from G1 were euthanized at D42, and all the remaining mice were euthanized at D60. Splenocytes were stimulated with synthesized peptides for 6 h before staining for flow cytometric detection. The mean percentage of epitope NP380-specific IFN-γ+CD8+ lymphocytes among CD8+ cells is shown in Fig. 3. Representative dot plots are shown in Fig. 3A. The box plots in Fig. 3B show the group means with statistical analysis results. In this study, NP380-specific IFN-γ+CD8+ lymphocytes were detected only in G2, G3, and G7. The mice in these three groups showed statistically higher cellular immune responses compared to G1. G7 had significantly higher cellular immunity than G2 (p<0.05). None of the other groups showed a response to NP380 stimulation. The IFN-γ-secreting cells were not detected in any of the groups upon stimulation of splenocytes with epitopes HA91, M2e, or NP366 (data not shown).

Flow cytometric analysis of the percentage of epitope NP380-specific IFN-γ+CD8+ lymphocytes among CD8+ cells. The numbers in the plots show the group mean percentages for G1, G2, G3, and G7 after in vitro stimulation of splenocytes with synthesized epitope NP380. None of the other groups showed a response to NP380 stimulation (data not shown). The representative dot plots acquired from flow cytometry are shown in

Epitope-specific humoral immune response
Epitope-specific humoral immunity was detected by ELISA, and the results are shown in Fig. 4. Mice in G3 seroconverted against epitope HA91 significantly (p<0.01) on D60 only after the protein boost at the third dose compared to the negative control group G1, but mice in G2 did not, even after the third dose of plasmid. However, mice in G4 showed a statistically significantly higher antibody response (p<0.001) on D42 after two doses of DNA vaccine. Antibody response against M2e was detectable on D42 in G2, and was nearly so in G3 after the second dose of DNA vaccine, and was significantly increased (p<0.05) on D60 after the third dose in G2 and G3. The mice in G5 had a significantly higher antibody response at D42 after the second dose of DNA vaccine. The mice in G2 and G3 showed some level of antibody against NP366 and NP380 at D60 after protein boost; however, no any antibody response was detected against these two epitopes in G6 and G7. For G1–G5, the serum collected after the mice were euthanized did not show any MN titer as determined by MN assay.
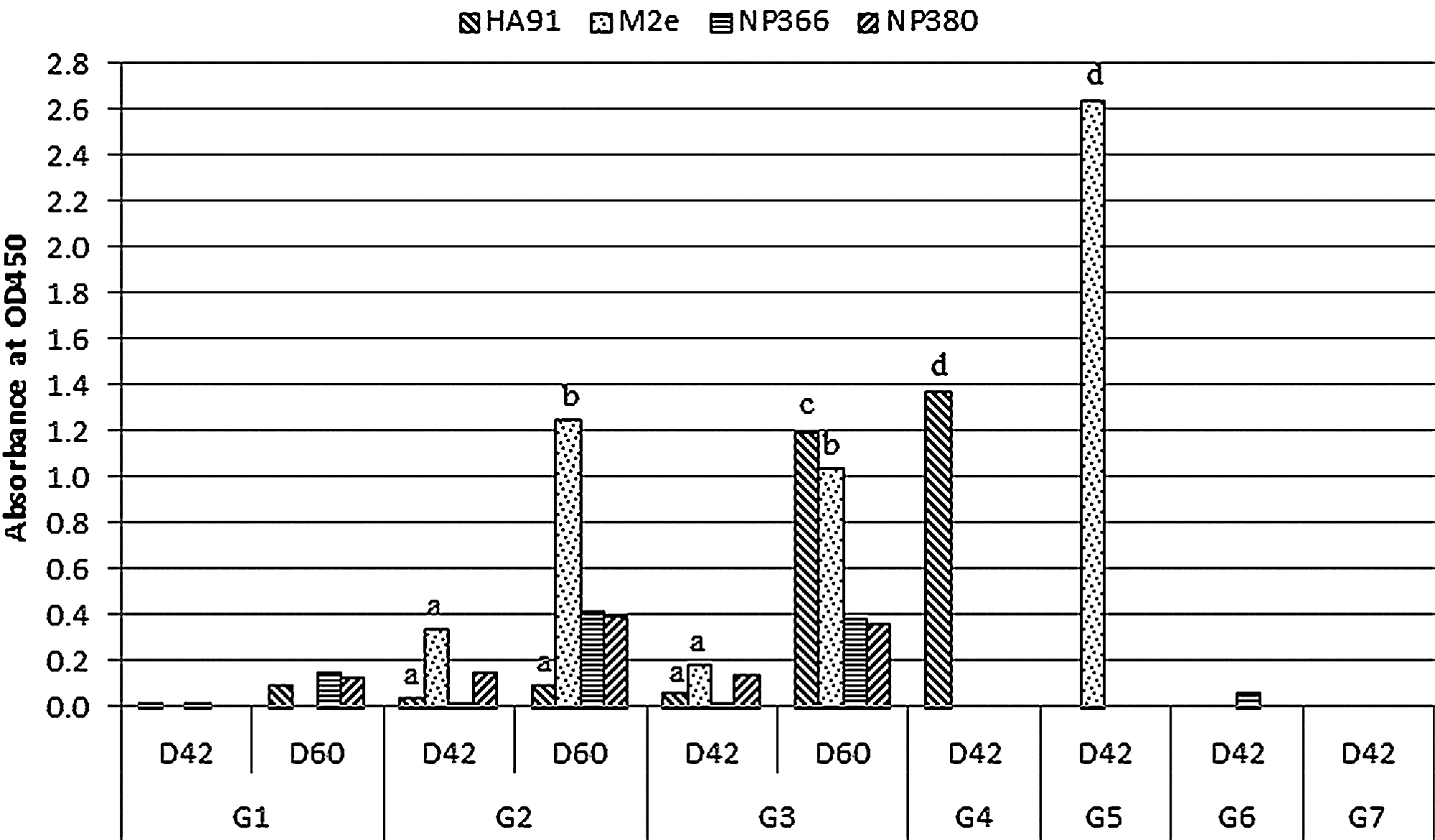
Epitope-specific antibody was detected by ELISA in serum at a dilution of 1:50 collected on study days 42 and 60. ELISA plates were coated with individual synthesized epitopes. A value >0.2 was determined to be positive. Statistical tests were conducted against the corresponding samples in G1 (a=p>0.05, b=p<0.05, c=p<0.01, d=p<0.001 compared to the sample in G1).
Discussion
Current influenza vaccines are not capable of conferring protection against emerging variants of even the same subtype as the vaccine strain. Herein, we explored the feasibility of an influenza DNA vaccine encoding the conserved B- and T-cell epitopes of the major subtypes of swine influenza virus.
In our study, we applied approaches in plasmid design for enhancing the expression and presentation of inserted epitopes. The in vitro transfection result measured by IFA (Fig. 1B) and flow cytometry (Fig. 1C) proved that the incorporation of VP22c to the N-terminal of epitopes dramatically increased the percentage of epitope-positive cells. Thus, only these plasmids were applied in the following mouse study. Different vaccination regimens were applied in the mouse vaccination study. In G2 and G3, a mixture of the four plasmids (pVP22cHA91, pVP22cM2e, pVP22cNP366, and pVP22cNP380) was applied, and compared to single-plasmid-vaccinated groups G4–G7. This was done purposely to demonstrate whether the combined injection of plasmids would be a feasible method in vaccine design compared to vaccination with a single plasmid. In addition, homologous and heterologous prime-boost regimens were implemented in this study to achieve a higher level of immune response.
The percentage of IFN-γ+CD8+ cells was measured by flow cytometry to indicate the level of cellular immunity upon vaccination. Upon in vitro peptide stimulation of splenocytes, only NP380-specific IFN-γ+CD8+ cells were detected, while no cellular immunity was detected against the other assumed T-cell epitope NP366 in the corresponding vaccinated groups, or against the two B-cell epitopes HA91 and M2e. Therefore, NP380 is a very immunogenic epitope in the construction in this study. This result shows that NP380 may be a good candidate for vaccine design, which is consistent with the other studies about this epitope (27 –30). The group mean percentage of IFN-γ+CD8+ cells against NP380 in G7 was statistically significantly higher than that in G2 (p<0.05), but was not significantly different from that in G3. The lower cellular immunity against NP380 seen in G2 and G3 may be related to the lower dose of the particular plasmid in G2 and G3 compared to G7. Unexpectedly, no cellular immunity was detected against NP366 in both combined and singly vaccinated groups. This is in conflict with what was shown in several studies (31 –33), in which NP366 was demonstrated to be a dominant epitope. It might be due to the way the plasmid was constructed in this study; the fusion protein of quadruplicated epitope NP366 was fused to the C-terminal of VP22c, and this might have changed the way that this epitope was processed and presented.
Epitope HA91 is confirmed to be immunogenic, as reported in the literature (27,28,30,34 –36). In our study, epitope-specific humoral immunity was detected positive against HA91 after two doses of DNA vaccine in G4, and after the third protein boost in G3, but not in G2 until the end of the study, as shown in Fig. 4. This indicates that the combined vaccination regimen does not result in competition among epitopes, since G3 showed a positive antibody response, the major reason for the lack of humoral immunity against HA91 in G2 might be due to the comparatively lower dose of DNA vaccination than what was in the single-plasmid vaccinated group G4. Another B-cell epitope used in this study, M2e, is very immunogenic, and is a potential means for developing universal vaccines (37 –43). In our study, M2e was more immunogenic than HA91, since mice in G2, G5, and nearly so in G3, seroconverted to this epitope after the second dose of DNA vaccine. After the third dose of either DNA in G2 or protein in G3, the antibody level against M2e was increased dramatically (p<0.05). Although they were still much lower than that in G5, the lower vaccine dose in G2 and G3 may explain this effect. Unexpectedly, mice in G2 and G3, after the third dose, showed a slightly positive antibody response to epitopes NP366 and NP380, compared to the negative results seen in G6 and G7. This indicates that the combined vaccination might have changed the way that these two T-cell epitopes were presented. The MN assay showed no neutralizing abilities of antibodies generated against HA91 and M2e. However, the epitope H3 HA91-108 proved able to reduce the viral load in the lungs against H3 virus infection when vaccinated in mice (34,44), although an MN assay was not conducted in those studies. Antibodies against M2e were shown to confer protection, as indicated by passive transfer experiments (38,42), and the M2e-specific antibodies bound to virus-infected cells rather than virus to neutralize infection (39). Antibody-dependent cell-mediated cytotoxicity (ADCC) is the only factor involved in protection, rather than C3-dependent cytotoxicity (39). So ADCC might also be the way epitope HA91 confers protection against viral infection.
Overall, the heterologous prime-boost strategy in G3 worked better for HA91 than the homologous vaccination strategy in G2, while they did not make a difference to M2e and NP380. The combined vaccination regimen in G2 and G3 showed a lower immune response to HA91, M2e, and NP380, compared to the corresponding single-plasmid vaccinated groups. The major reason is likely the dose restriction in the former groups.
The ideal DNA-epitope vaccine able to induce wide-spectrum immunity can be achieved only through vaccination with the appropriate combination of both B- and T-cell epitopes. Many epitopes have been studied in the literature for influenza viruses. In this study, SIV-derived epitopes were inserted into modified DNA plasmids and stimulated epitope-specific immunity successfully via intramuscular administration into mice. With the established DNA-epitope vaccine platform described in this study, more epitopes can be screened for their immunogenicity, followed by testing the best combinations of plasmids. To overcome the dose restriction needed in combined vaccination, the fusion of multiple epitopes in one plasmid represents a feasible approach for future studies.
Footnotes
Acknowledgments
We thank the Showalter Trust Fund for funding of this study. We also thank Virginie Lazar for assistance with designing the primers used in this study.
Authors Disclosure Statement
No competing financial interests exist.