
Abstract
Human cytomegalovirus (HCMV) activation and elevated levels of vascular endothelial growth factor (VEGF) have been found to be associated with transplant rejection. However, information is lacking about whether elevated levels of this multifunctional factor are directly due to viral activation, or if they derive from impurities within the culture supernatant of infected cell cultures. We used purified as well as unpurified viral stocks to infect human fibroblasts in vitro and applied PCR, Western blot, ELISA, and immunofluorescence staining to investigate the expression of VEGF and its receptors. Our data suggest that HCMV infection triggers an early and sustained induction of VEGF and kinase insert domain receptor (KDR) mRNAs, whereas transcript levels of FLT-1 remain unchanged by viral infection. Analysis of the extracellular VEGF and cellular KDR protein expression after infection with purified and unpurified HCMV strains AD169 and TOLEDO showed, in clear contrast to UV-inactivated virus preparations, an increased release of VEGF and KDR proteins. In addition, immunofluorescence revealed that HCMV infection was also accompanied by a profound increase in intracellular VEGF and KDR levels. These results confirm that active HCMV infection is required to induce VEGF and the most important VEGF receptor KDR, and that the upregulation of VEGF and KDR are a direct viral effect and not a secondary effect mediated by inflammatory cytokines within the supernatant. The HCMV-dependent upregulation of VEGF and KDR contributes to the theory that viral-induced immune mediators play a key role in transplant rejection.
Introduction
One of the major effects of HCMV is its ability to modulate the immune system of the host. CMV infection can cause an autoimmune phenomena in patients as well as immunosuppressive effects (31). However, a hallmark of HCMV infection is its induction of an extensive immune response. This can occur via infection and following activation of several cells. For instance, HCMV can activate endothelial cells, smooth muscle cells, fibroblasts, monocytes/macrophages, and dendritic cells, leading to a wide range of immune responses (31), which encompass the release of multiple inflammatory effectors (such as chemokines and cytokines), as well as the recruitment of lymphocytes and monocytes (29). Diverse immune responses contribute to the vascular damage, which is a key factor in allograft rejection following CMV infection. Recently, a link between enhanced levels of the immune modulator macrophage migration inhibitory factor (MIF) and renal graft rejection has been established (15). Our study on the role of MIF during HCMV infection in fibroblasts provided the first evidence for enhanced levels of MIF transcripts and protein expression following HCMV infection (1). Reinhardt et al. further demonstrated an upregulation of vascular endothelial growth factor (VEGF) by HCMV in fibroblasts (22). VEGF levels were also found to be elevated in graft rejection, and post-operative trapping of VEGF could lead to improved long-term graft survival (6).
VEGF is a 46-kDa heparin-binding growth factor, and has long been established as the most important angiogenetic factor. Five isoforms derive from alternative splicing from a single copy of the VEGF gene (8,17). These isoforms differ in their molecular mass and biological activities and can bind to three different receptors, VEGFR1 (Flt-1), VEGFR2 (Flk-1/KDR), and VEGFR3 (Flt-4). Under physiological conditions, VEGF acts as a potent proangiogenetic factor by stimulating proliferation and migration of endothelial cells and preventing them from undergoing apoptosis and senescence (30). VEGF expression is markedly increased in response to hypoxia (26). Under pathological conditions, VEGF overexpression can lead to several clinical issues; for instance, its expression can be increased following oncogene activation, resulting in tumor angiogenesis (20). VEGF can also act as a proinflammatory cytokine. It mediates vascular permeability, enhances induction of adhesion molecules on endothelial cells, and acts as a leukocyte attractant (21). These features can lead to arteriosclerotic plaque formation (5), and have also been found to contribute to cardiac allograft arteriosclerosis and the subsequent graft rejection (16).
Since the role of HCMV infection in the regulation of VEGF and its receptors has not been evaluated, we analyzed the effect of two different HCMV strains, AD169 and TOLEDO, on the regulation of VEGF, VEGFR-1 (Flt-1), and VEGFR-2 (KDR), in human foreskin fibroblast (HFF) cells.
Methods
Cells
HFF cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 2 mM glutamine, 50 U/mL penicillin, and 50 μg/mL streptomycin at 37°C and 5% CO2. For kinetics, confluent HFF cells between passages 20 and 25 were washed with PBS and maintained in FCS-free DMEM supplemented with 2 mM glutamine, 50 U/mL penicillin, and 50 μg/mL streptomycin. HFF cells were infected with active purified as well as unpurified HCMV, UV-inactivated HCMV, and HCMV, in the presence of 200 μg/mL phosphonoacetic acid (PAA). Monolayer cultures (1.5–5×106 cells each) were harvested by scraping after collection of the medium.
Virus strains
The laboratory strains AD169 and TOLEDO of HCMV were used. For experimental infection a multiplicity of infection (MOI) of about 3 was used. Virus titrations were performed by the end-point dilution method using indirect immunofluorescence for the detection of immediate early HCMV antigen with a commercial mAb (18).
HCMV was inactivated by exposure to a 254-nm germicidal UV lamp at a distance of 8 cm for 60 min (HCMV-UV). The effect of the UV treatment to produce noninfectious virus was confirmed by the failure to produce a characteristic cytopathic effect (CPE), as this had been achieved with infectious virus preparations. To inhibit HCMV DNA synthesis, infected cells were treated continuously with 200 μg/mL PAA (13) starting immediately after virus absorption. In addition, virus particles were purified by velocity centrifugation in a sucrose gradient as previously described (28). Briefly, 27 mL of an infected cell supernatant prepared as described above was underlaid with a 5-mL sucrose cushion containing 20% sucrose, 50 mM Tris-HCl (pH 7.2), and 1 mM MgCl2. Virions were pelleted by centrifugation at 24,000×g for 16 h at 4°C. The pellets were resuspended in DMEM to produce a purified virus stock.
Reverse transcriptase polymerase chain reaction (RT-PCR)
RT-PCR was used to analyze transcription of VEGF, FLT-1, KDR, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as control. RNA was extracted from HFF by using Trizol reagent (GibcoBRL, Karlsruhe, Germany) in accordance with the manufacturer's instructions. First, 1 μg total RNA was reverse transcribed with 40 U of Superscript II (GibcoBRL) and 1 μg oligo-(dT)18 in a total volume of 20 μL following the manufacturer's protocol. The following intron-spanning primer pairs were used to amplify cDNAs: hVEGF (587 bp): 5′ primer: 5′-TGG ATC CAT GAA CTT TCT GCT-3′, 3′ primer: 5′-GGA ATT CAC CGC CTC GGC TTG TC-3′; hGAPDH (540 bp): 5′ primer: 5′-CGT CTT CAC CAC CAT GGA GA-3′, 3′ primer: 5′-CGG CCA TCA CGC CAC AGT TT-3′; hFLT-1 (480 bp): 5′ primer: 5′-GCT CAC CAT GGT CAG CTA CT-3′, 3′ primer: 5′-GTG TAT AAT TTC GGG GAT TTC-3′; and hKDR (540 bp): 5′ primer: 5′-GTC ATT TAT GTC TAT GTT CAA G-3′, 3′ primer: 5′-CCC ACT GGA TGC TGC ACA GGT-3′.
Next, 1-μL aliquots of cDNA were amplified with a PCR system 2400 thermocycler (PerkinElmer, Weiterstadt, Germany) for VEGF using the primers described above with the following cycling program: denaturation for 45 sec at 95°C, annealing for 60 sec at 60°C, and extension for 60 sec at 72°C. PCR products were analyzed for VEGF, FLT-1, KDR (35 cycles), and GAPDH (30 cycles), after amplification and gel electrophoresis in 1% agarose gels. Semiquantitative determination was achieved by digitization of gels with a Polaroid video system (Rothaar & Schroeder, Heidelberg, Germany), following subsequent densitometric evaluation with the Gelscan 4.0 professional program (LTF/Bio SciTec, Landau/Frankfurt, Germany).
DNA-sequence analysis
For sequencing, PCR amplification products were gel-purified (QIAquick Gel Extraction Kit; Qiagen, Hilden, Germany). Direct sequencing of PCR products was performed by cycle sequencing in both directions with ABI PRISM Big Dye Terminator chemistry (Perkin-Elmer), followed by electrophoresis on a Perkin-Elmer ABI-377 automated sequencer.
VEGF and KDR Western blot analysis
Supernatants were collected from mock- or HCMV-infected cells at the times indicated in the results section and figure legends. To inhibit CMV DNA synthesis, infected cells were treated continuously with 200 μg/mL PAA (13) starting immediately after virus adsorption. Upon sampling, protease inhibitor cocktail (Boehringer, Mannheim, Germany) was added and the supernatants were clarified by centrifugation at 2000×g for 5 min at 4°C and 2-mL aliquots were concentrated in Centricon-10 concentrators (Amicon, Beverly, MA) according to the operating manual. The concentrated samples were kept frozen at −80°C until tested. Intracellular protein was isolated by the Trizol method as outlined in the manufacturer's manual. Protein content of the cell lysates was quantified using the BCA kit (Pierce, Rockford, IL). The concentrated supernatants and intracellular protein were separated by SDS-PAGE and blotted onto Optitran BA-S 83 nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany), using Nu PAGE pre-cast 4–12% bis-tris polyacrylamide gels (Novex, San Diego, CA), according to the manufacturer's instructions. Western blotting was performed according to the instructions of the Super Signal Ultra Western blotting kit (Pierce). Briefly, the blots were incubated with goat anti-human VEGF IgG and rabbit anti-human KDR IgG. The membranes were washed four times with PBS containing 0.5% Tween-20, and were then incubated for 1 h with horseradish-peroxidase conjugated secondary antibody (rabbit anti-goat IgG [VEGF] and goat anti-rabbit IgG; dilution 1:250,000). The blots were washed 4×10 min and incubated for 5 min in Super Signal substrate working solution (Pierce), and exposed to Bio Max MR autoradiographic film (Kodak, Rochester, NY). The films were processed in an Automax automatic developing machine (MS Laborgeraete, Heidelberg, Germany). For further semiquantitative determination, the developed films were densitometrically evaluated with the Gelscan 4.0 Professional Program.
VEGF ELISA
For measurement of VEGF protein, supernatants from HCMV-infected HFF cells were analyzed by sandwich ELISA, applying a monoclonal anti-human VEGF capture antibody, biotinylated goat anti-human VEGF IgG detection antibody (R&D Systems, Wiesbaden, Germany), and purified human VEGF as standard.
VEGF and KDR immunofluorescence assays
Cells were permeabilized and fixed by acetone/methanol (1:1) for 30 min at 4°C. Visualization of intracellular VEGF and KDR protein expression was carried out by indirect immunofluorescence with goat anti-human IgG (VEGF), rabbit anti-human IgG (KDR), and a secondary fluorescein isothiocyanate (FITC)-labeled rabbit anti-goat IgG and goat anti-rabbit antibody (Dako A/S, Glostrup, Denmark) for 30 min at 4°C. For documentation, a Zeiss Axiophot microscope and Spot camera were used.
Results
Effect of HCMV infection on VEGF, FLT-1, and KDR mRNA expression in human HFF cells
To examine any possible induction of VEGF and its receptors FLT-1 and KDR in HFF cells following HCMV infection, we first studied the corresponding expression of mRNAs. In infected fibroblasts VEGF and KDR steady-state mRNA levels were detected at the beginning of the incubation period, and reached maximum levels at the end of the infection course at 96 h (Fig. 1A and B), whereas transcripts for FLT-1 and GAPDH remained unchanged during viral infection (Fig. 1C and D). Densitometric evaluation of PCR products confirmed and extended our results (Fig. 1E). Several studies have shown the VEGF mRNA121 isoform to be the most highly expressed form in human tissues and tumors, whereas expression of VEGF165, VEGF189, and VEGF206 is variable. That result was also confirmed by our study. Therefore, we decided to quantify only the induction of the VEGF189 and VEGF165 mRNA species.
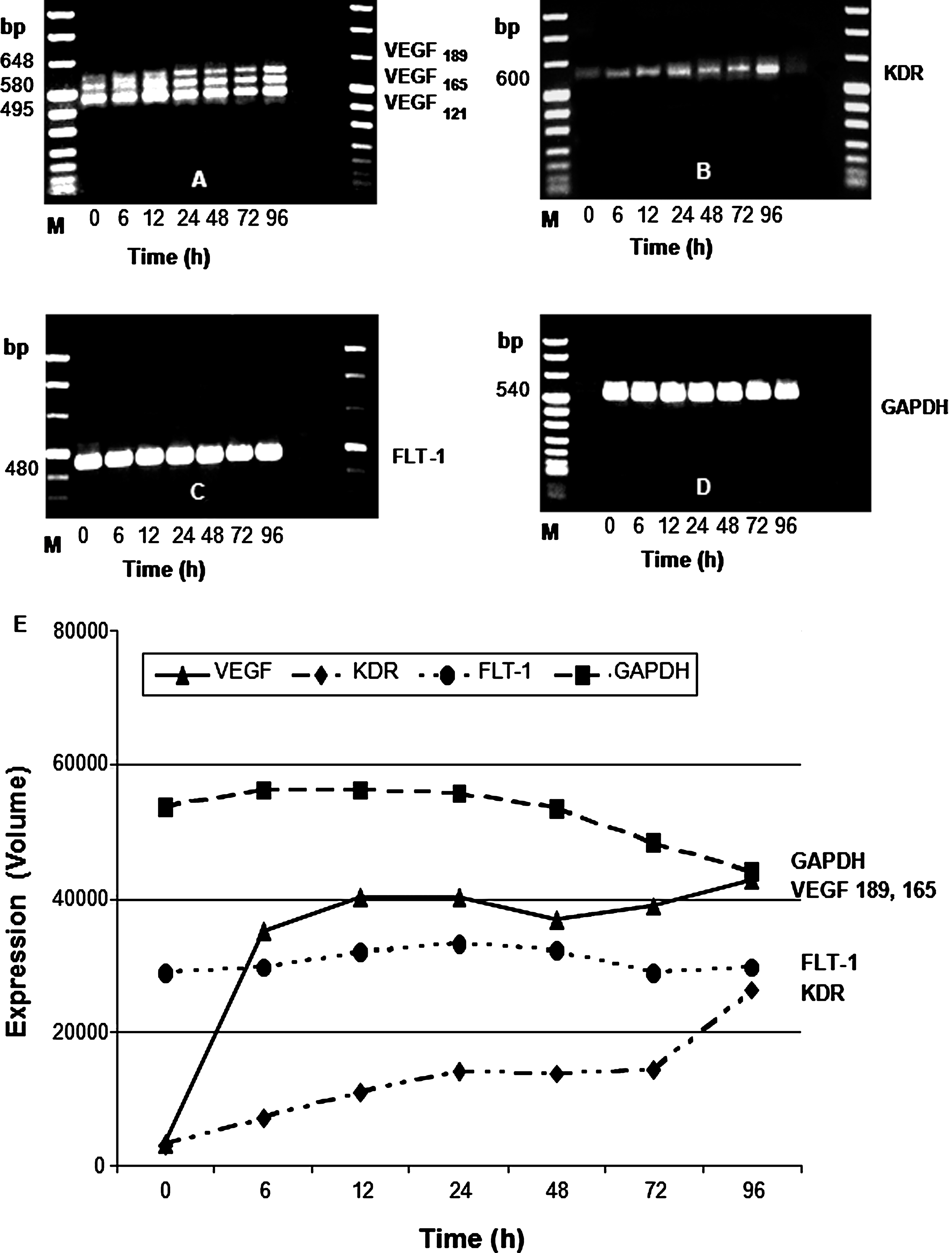
Expression of VEGF (
The experiment described above used an unpurified virus stock (i.e., an infected cell supernatant), as the source of virus. Such a virus stock almost certainly contains various biological compounds, cytokines, and other modulating factors, that have been described during the course of viral infection. These factors in the virus stock may be responsible for, or may contribute to, the observed induction of VEGF and KDR mRNA. To distinguish between direct induction of VEGF and KDR by the virus and induction by a contaminating factor, we purified the virus stock.
HCMV (AD169 and TOLEDO) induce VEGF protein expression in human HFF cells
To examine whether elevated VEGF mRNA levels result in enhanced VEGF production and thus higher extracellular VEGF contents, we analyzed supernatants of infected HFF cells using Western blot. Following treatment with either purified AD169, unpurified AD169, or UV-inactivated AD169, we observed enhanced levels of VEGF protein with unpurified as well as with gradient-purified AD169 virus preparation upon viral infection. In comparison to the infection with purified AD169 virus, the infection with unpurified AD169 virus led to a notable upregulation of VEGF after 12 h of treatment (Fig. 2B and C). The incubation with the supernatants of the UV-inactivated virus failed to induce enhanced levels of VEGF in HFF cells (Fig. 2D). Additionally, we analyzed extracellular VEGF protein expression after unpurified or gradient-purified TOLEDO virus preparation, since TOLEDO virus is more infectious than AD169 virus. We also detected an upregulation of VEGF protein with the unpurified or gradient-purified virus (Fig. 2E and F). The comparison between AD169 and TOLEDO infections revealed that the VEGF expression occurred earlier and was more pronounced with the purified and unpurified TOLEDO virus strains (Fig. 2).
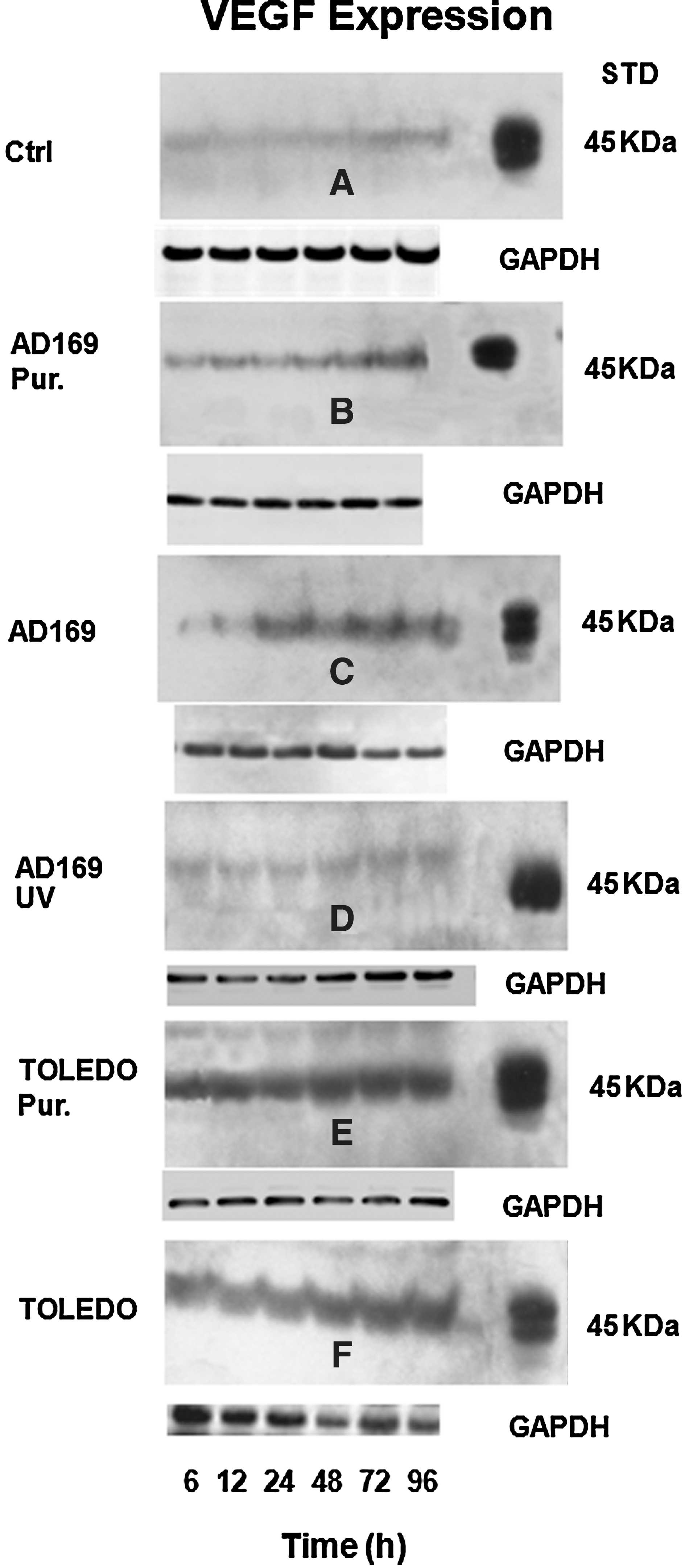
VEGF content in supernatants from HCMV-infected fibroblasts. The cells were infected with purified AD169 (AD169 Pur.;
To further validate our findings of an upregulation of VEGF production in HFF cells following virus infection, we applied a VEGF-specific sandwich ELISA. Using supernatants of HCMV-infected cells, HCMV-UV treated cells, and non-treated control cells, only HMCV-infected cells showed increasing levels of VEGF protein in the supernatant. In contrast, low levels of VEGF were detected when PAA- or UV-inactivated virus was added to the cell cultures (Fig. 3). PAA, which inhibits HCMV DNA synthesis, was applied to determine whether the HCMV-dependent induction of VEGF protein is a late event of the viral cycle.
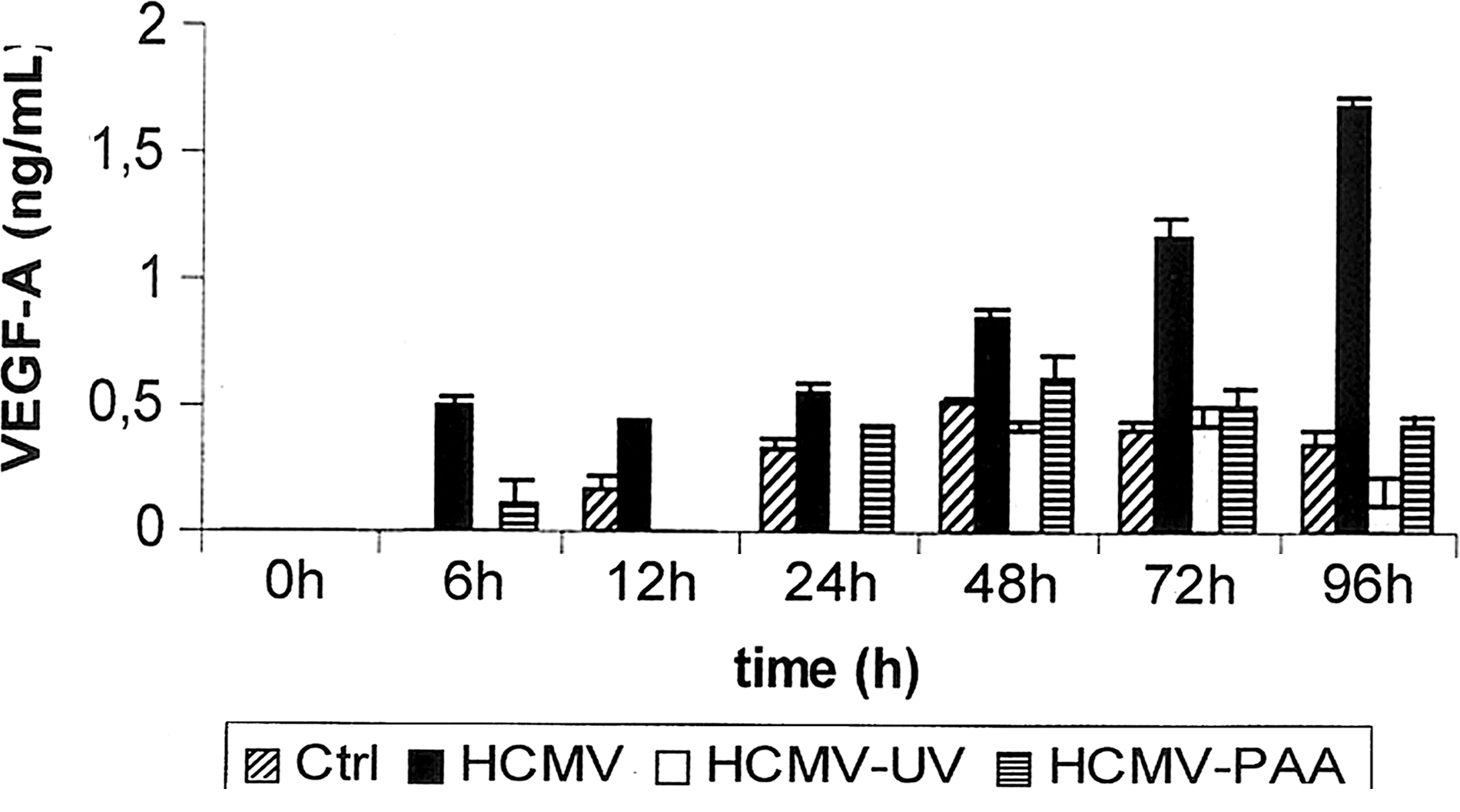
VEGF secretion by human fibroblasts. Extracellular VEGF production was analyzed by sandwich ELISA of supernatants collected at the indicated time points from uninfected fibroblasts (Ctrl), from HCMV-infected cells (HCMV), from fibroblasts incubated with UV-inactivated virus (HCMV-UV), and from HCMV-infected cells that were incubated in the presence of PAA (HCMV-PAA). Measurements were performed in triplicate cultures; the data shown are the mean±SD of one representative experiment out of a group of four separate experiments with similar results.
HCMV (AD169 and TOLEDO) induce VEGFR-2 (KDR) in human HFF cells
We further questioned whether the elevated KDR RNA levels shown in Fig. 1 also lead to increasing KDR protein levels in HFF cell supernatants following HCMV infection.
Whereas infection with purified AD169 virus induced KDR expression after 48 h (Fig. 4B), unpurified AD169 virus led to an early rise in KDR protein levels after only 6 h (Fig. 4C). However, both purified and unpurified TOLEDO virus infection caused markedly elevated KDR levels after 6 h (Fig. 4E and F). Neither non-treated (control) nor UV-inactivated AD169-treated HFF cells showed enhanced KDR protein levels in supernatants (Fig. 4A and D). Densitometric quantification data are shown in Fig. 4G, and show that infection with unpurified and gradient-purified virus of AD169 and TOLEDO led to an increase of VEGF protein levels in HFF cells.

Kinetics of KDR production in HCMV-infected human fibroblasts. The cells were infected with purified AD169 (AD169 Pur.;
Detection of cellular VEGF and KDR in human HFF cells following HCMV infection
Using indirect immunofluorescence, we were able to show elevated cellular VEGF and KDR levels following HCMV infection (Fig. 5A and B). Whereas control HFF cells did not show any cellular VEGF and KDR localization, VEGF and KDR expression in HFF-infected cells slowly increased after 12–24 h, leading to a distinct rise toward the end of the infection course at 96 h. This virus-dependent induction of VEGF could be the reason for the elevated KDR protein expression seen in human HFF cells, which was also confirmed by the Western blots obtained from cell lysates (Fig. 4).
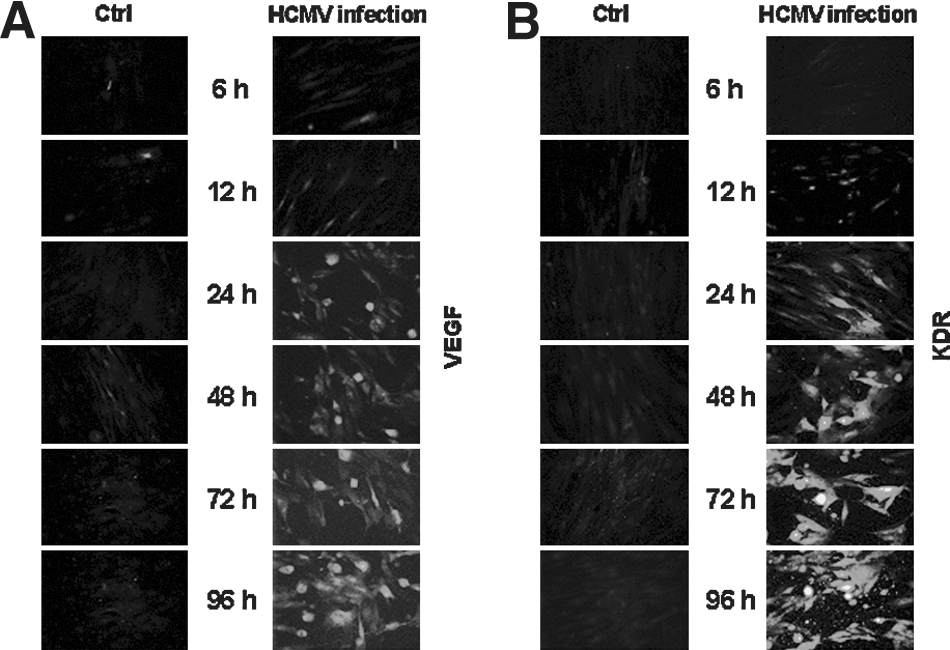
Immunofluorescence detection of VEGF and KDR in HCMV-infected human fibroblasts. Intracellular VEGF (
Discussion
Previous studies have shown HCMV infections to be frequently associated with acute and chronic transplant rejection (4,10). Following graft transplantation, HCMV infection can cause a variety of effects, such as damage to endothelial cells, the release of proinflammatory cytokines and chemokines, the recruitment of leukocytes, and the upregulation of adhesion molecules on endothelial cells (31). Thus far, effective antiviral chemotherapy has been shown to be of some benefit to prevent the progress of CMV disease following transplantation, but it has been of limited success in improving graft survival (14,27).
Elevated levels of VEGF and its corresponding receptors has also been linked to enhanced graft rejection (23). VEGF is a strong angiogenetic effector, leading to the proliferation of various cells, such as endothelial cells (2,7). However, VEGF can also act as a proinflammatory cytokine causing pathological conditions. The effects of VEGF are mediated through binding to one of its three receptors: VEGFR-1 (Flt-1), VEGFR-2 (Flk-1/KDR), and VEGFR-3 (Flt-4). VEGFR-3 is predominantly expressed on lymphatic endothelial cells and may play a critical role in pathological lymphangiogenesis following graft transplantation (3). VEGF-1 and VEGFR-2 are mainly found on vascular endothelial cells and mediate downstream signals, resulting in cell proliferation, migration, permeability, and survival, finally leading to vasculogenesis and angiogenesis (12). However, VEGFR-2 is found to be the key regulator in these conditions, whereas VEGFR-1 might contribute to and modulate VEGFR-2 signaling. VEGF is likely to upregulate VEGFR-2 levels in an autocrine fashion (25,32). It has recently been shown that VEGFR-1 and VEGFR-2 contribute to pathological conditions following cardiac allograft rejection, such as excessive inflammation and arteriosclerosis (19).
In our study, we determined whether HCMV infection could regulate VEGF, Flt-1, and KDR expression and protein levels in HFF cells. We found that the consistently high levels of FLT-1 in HFF cells were not altered during the course of viral infection. In striking contrast, VEGF (VEGF189, VEGF165, and VEGF121) and KDR mRNA levels were induced in HFF cells following HCMV infection. By using UV-inactivated HCMV as a control, we confirmed that this upregulation requires active HCMV. UV-inactivated HCMV did not display a cytopathic effect in HFF cells or induce mRNA levels of these factors. The increase of VEGF and KDR transcripts during the course of AD169 and TOLEDO infection was accompanied by a similar increase in protein expression of these factors. We demonstrated that both purified and non-purified viruses could increase the expression levels of KDR and VEGF, which confirmed that this effect was not due to a contaminating factor within the supernatants. The inactivation of the strains AD169 and TOLEDO by UV irradiation completely abrogated the induction of VEGF and KDR protein expression in HFF cells, which further indicates that active viral infection appears to be required for the induction of VEGF and KDR expression. These data were confirmed by immunostaining. We provided clear evidence of HCMV-dependent induction of VEGF and its most important receptor KDR. We further suggest that VEGF expression is induced by HCMV, whereby VEGF itself might upregulate its receptor KDR in HFF cells. This raises the possibility of the autocrine stimulation of VEGF via its receptor KDR.
Given the fact that both HCMV infection and increased VEGF levels were independently found to be linked to a higher risk of graft rejection, our findings of an HCMV-dependent upregulation of VEGF and its most important receptor KDR could provide further insights into the mechanism of allograft rejection. VEGF overexpression following graft transplantation was found to promote lymphangiogenesis across the wound edges by enhanced leukocyte trafficking between the allograft and secondary lymphoid organs (3,19). This process is mainly mediated through VEGFR-3 signaling, and can be considered a first step in the process of allograft rejection (3,6). By binding to its receptors VEGFR-1 and predominantly VEGFR-2, VEGF can further aggravate this immunological process by directly enhancing the recruitment of monocytes and T cells, endothelial chemokine production, and the induction of endothelial adhesion molecules (21). It can further promote the migration and upregulation of smooth muscle cells. These features are likely to provoke vascular damage and vessel narrowing, causing an arteriosclerotic pattern in graft-associated vessels, and ultimately leading to graft rejection (19).
Conclusions
Taken together, our experiments demonstrate that HCMV promotes the production of VEGF and KDR in primary human fibroblasts. HCMV-dependent increases of VEGF and KDR could directly contribute to inflammatory processes that eventually lead to graft rejection.
Footnotes
Acknowledgments
The authors thank Drs. Markus Eickmann, Amirreza Vahhabzdeh, and Andreas Kaufmann for technical assistance.
Author Disclosure Statement
No competing financial interests exist.