
Abstract
Selecting for antibodies against specific cell-surface proteins is a difficult task due to many unrelated proteins that are expressed on the cell surface. Here, we describe a method to screen antibody-presenting phage libraries against native cell-surface proteins. We applied this method to isolate antibodies that selectively recognize CCR5, which is the major co-receptor for HIV entry (consequently, playing a pivotal role in HIV transmission and pathogenesis). We employed a phage screening strategy by using cells that co-express GFP and CCR5, along with an excess of control cells that do not express these proteins (and are otherwise identical to the CCR5-expressing cells). These control cells are intended to remove most of the phages that bind the cells nonspecifically; thus leading to an enrichment of the phages presenting anti-CCR5-specific antibodies. Subsequently, the CCR5-presenting cells were quantitatively sorted by flow cytometry, and the bound phages were eluted, amplified, and used for further successive selection rounds. Several different clones of human single-chain Fv antibodies that interact with CCR5-expressing cells were identified. The most specific monoclonal antibody was converted to a full-length IgG and bound the second extracellular loop of CCR5. The experimental approach presented herein for screening for CCR5-specific antibodies can be applicable to screen antibody-presenting phage libraries against any cell-surface expressed protein of interest.
Introduction
CCR5 is the major co-receptor of human immunodeficiency virus type-1 (HIV-1) and of HIV-2, thus playing a pivotal role in HIV transmission and pathogenesis (13,18). Consequently, it has been studied intensively as a potential target for drugs effective against both HIV-1 and HIV-2 infections (32,36,42,59,61). So far, a number of small-molecule CCR5 antagonists have been identified and demonstrated potent antiviral effects both in cell culture and in clinical trials (36,39,40,42,59). Furthermore, several of anti-human CCR5 mAbs were discovered and their therapeutic use is under investigation in preclinical or clinical trials (30,31).
CCR5 belongs to the A family of the G protein-coupled receptors (GPCR) with characteristic seven-trans-membrane domains (1,14,16). It has an N-terminal exo-domain and three extra cellular loops (ECLs). Therefore, this protein can offer multiple extracellular epitopes for recognition by specific Abs. Like most GPCRs, CCR5 is naturally expressed on the cell surface at low levels (34,37). Nevertheless, the recombinant expression of GPCRs in bacterial, yeast, or insect cells can result in protein misfolding and aggregation (55). Furthermore, CCR5 requires post-translational modifications and hence, the recombinant is likely to differ from the natural protein, when expressed in non-mammalian cells (19). On the other hand, direct purification of the naturally-expressed CCR5 and other GPCRs from mammalian cell membranes may lead to irreversible protein misfolding and denaturation. In this case, the screening for potential Abs with purified CCR5 may result in Abs that also recognize the intracellular domains of the protein, which are not accessible for binding of Abs when the protein is naturally expressed on the cell surface. Therefore, it is less practical to use the purified CCR5 protein for screening assays. An additional solution for isolating Abs against integral membrane proteins is using synthetic peptides derived from sequences of the protein's outer-membrane domains. This approach suffers from many limitations and in most cases, peptide-specific Abs fail to recognize the natural protein target (27).
In the study described here, we present a general approach for screening phage libraries using flow cytometry, in order to isolate molecules that interact specifically with the extracellular epitopes of membrane proteins. In this strategy, we have co-expressed on the target cells the plasma membrane integral protein, CCR5, along with the intracellular marker green fluorescent protein (GFP). The cellular expression of recombinant CCR5 achieves two goals. First, the cells display the CCR5 protein at higher density than in the naturally-expressing cells, thus increasing the sensitivity of the phage screening procedure. Second, similar cells that do not present CCR5 and GFP serve as ideal control cells for removing nonspecific binders. Since these control cells are used in excess, incubating the phages with this mixed cell population will result in a preferential binding of the CCR5-specific phages to the positive, CCR5 and GFP-expressing, cells. GFP expression allows a quantitative separation of the positive cells by cell sorting, thus isolating the specific phages that bind only these cells. Following this procedure, we succeeded in isolating five novel human anti CCR5-specific Abs from phage libraries that display human single chain fragment variable (ScFv) Abs. One of the ScFvs, designated A2 mAb, showed the highest affinity towards CCR5. Therefore, it was produced also as the IgG format for achieving a better affinity towards the antigen.
Materials and Methods
Cells
The cell lines used were: human embryonic kidney cells 293T (17), mouse fibroblast cells expressing human CD4, 3T3.T4 (15), mouse fibroblast cells expressing both human CD4 and CCR5 epitopes, 3T3.T4.CCR5 (that carries also a selective puromycin marker) (15), canine thymocytes expressing the CXCR4 and CCR5 GPCRs, Cf2Th-CXCR4 (3) and Cf2Th/syn CCR5 (46), respectively. The latter four lines were a generous gift from the AIDS Research and Reference Reagent Program. The cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) high glucose medium, complemented with 10% heat-inactivated fetal calf serum (FCS), 2 mM L-glutamine, 100 units penicillin, and 100 μg/mL streptomycin (pen-strep).
Antibodies
The different Abs used in this study were: goat anti-mouse IgG, conjugated to a phycoerythrin (PE) (Jackson Immunoresearch Laboratories); mouse anti-CCR5 IgG mAb–2D7 (BD Biosciences); biotin-conjugated rabbit Abs against the bacteriophage fd protein (Sigma); streptavidin-R-phycoerythrin (PE) conjugated goat-anti rabbit IgG (H+L) (Sigma); mouse anti-c-Myc mAb IgG - 9E10 (Santa Cruz); horseradish peroxidase (HRP)-conjugated anti-mouse IgG (Santa Cruz); fluorescein isothiocyanate (FITC)-labeled goat anti-mouse IgG Ab (Jackson Immunoresearch Laboratories).
HIV-1 pseudovirus preparation and infection of the CCR5-expressing cells
For stable expression of GFP in 3T3.T4.CCR5 cells, pseudoviruses (PVs) were prepared in human kidney 293T cells, as previously described by us (24,25). The 293T cells were plated on 10-cm plates, and on the following day they were co-transfected with three plasmids: 10 μg of pCMVΔ8.2Gag-pol, which encodes for HIV-1 proteins (except for Env), 10 μg of pHRCMV-GFP (that is transcribed into mRNA containing encapsidation signal and the GFP coding sequence), and 3 μg of pVSV-G, which supplies a vesicular stomatitis virus (VSV) envelope protein (thus, allowing infection of a wide variety of cells). Chloroquine was added to the medium prior to transfection (to a final concentration of 25 μM). 48 hours post transfection, the supernatant, containing the PVs, was collected. Filtered HIV-1 PVs were assayed for the p24 Gag antigen by ELISA, using the HIV-1 p24 antigen-capture assay kit (from AIDS Vaccine Program, NCI, Frederick MD). The 3T3.T4.CCR5 cells were infected with 4.5 ng of p24 Ag PVs, in the presence of 5.6 μg/mL polybrene. The cells were collected 72 h post infection and sorted using a FACSAria cell sorter (BD Biosciences). This sorting was performed according to the cells' green fluorescence, as well as the high expression of CCR5. This was done using the commercial mouse anti-CCR5 mAb–2D7, followed by goat anti-mouse IgG, conjugated to a phycoerythrin (PE) marker (Jackson Immunoresearch Laboratories). After each sorting cycle, the cells were recovered by growth in DMEM containing 3 μg/mL puromycin to avoid genetic reversions. The finally-isolated cells were designated 3T3.T4.CCR5-GFP.
Phage display libraries
We have used the Metha1 and Metha2 libraries that disply ScFv Abs on the surface of the phages by fusing each ScFv to the pIII protein of the M13 bacteriophage. The two libraries were constructed from B cells, isolated from the peripheral blood of non-immunized human volunteers. The Metha1 and Metha2 libraries, containing 1.2×1010 and 1.5×1010 members, respectively, were constructed through the random assembly of VH genes with V-kappa and V-lambda genes (44). The phages from a mixture of the two libraries were recovered from E. coli TG1 bacteria stocks that release the phage particles after superinfection with the helper phage. The mixture was grown in 2xTY medium by shaking at 37°C until the 600nm optical density of the culture reached ∼0.5 (that equals to about 108 bacteria/mL). Then, 10 mL from the culture (∼109 bacteria) was infected by the helper phage, VCS-M13, at a phage/E. coli ratio of 20:1 (2×1010 plaque forming units (pfu)/mL). The helper phage, which provides the proteins required for the assembly of viable phage particles, was previously titered. Infection with helper phage and the rescue of phage particles was performed as described (4).
Selection of the libraries
The generated 3T3.T4.CCR5-GFP cells were used for affinity-selecting the Metha1 and Metha2 phage libraries. Affinity selections were performed as follows: On the first round of selection, approximately 2×107 3T3.T4.CCR5-GFP cells were blocked by incubating for 1 h in 10 mL of saturation buffer (SB) that is composed of 5% bovine serum albumin (BSA) in phosphate-buffered saline (PBS). Cells were then centrifuged at 210 g for 6 min and re-suspended with 1011–1012 pfu of phage, diluted in 1 mL PBS, containing 2% BSA. The cells and the phages were incubated at room temperature for 1 h, followed by washing ten times with 10 mL SB and twice with 10 mL PBS. Elution of phages was performed by re-suspending the cells in 1 mL of 0.1 M triethylamine for 10 min. Then, 250 μL of 2 M Tris-HCl (pH 7.4) was added to neutralize the pH of the eluate. This procedure lyses the cells without damaging the phages. The solution was then used directly to infect E. coli TG1 bacteria. Phage enrichment was performed with helper phage, as described above. From the second round of selection, 1012 pfu phages were incubated for 1 h at room temperature with 8×107 control 3T3.T4 cells, along with 2×107 3T3.T4.CCR5-GFP cells (yielding a 4:1 ratio, respectively). After incubation, the cells were washed again, and sorted by flow cytometry to isolate CCR5-expressing cells that co-express GFP, as described above. The sorted cells were collected and phages were eluted by re-suspending in 1 mL of 0.1 M triethylamine. After neutralization, the resultant supernatant was used again for bacterial infection. The same procedure was also conducted in the third and fourth rounds of infection.
Flow cytometry analyses of phages and antibodies
The 3T3.T4 and 3T3.T4.CCR5 cells were suspended in SB and incubated for 1 h with 500 μL of phage-containing solution (containing ∼1011 pfu) or with 500 μL of the bacterial peri-plasmic supernatant that contains the soluble ScFv antibody preparations (prepared as described below). The cells were washed with 10 mL of SB containing 5% FCS (instead of 5% BSA) in PBS. Cells were then centrifuged and incubated for 30 min with the appropriate Abs. The cells, mixed with the phages, were treated with biotin-conjugated rabbit Abs against the bacteriophage fd protein (diluted 1:500 in SB), while the cells that were incubated with the ScFvs were treated with 1 μL anti-c-Myc mAb (as the c-Myc epitope is part of the C-terminus of all presented ScFvs). Cells were washed as described above, and then incubated with streptavidin-PE conjugated-goat anti-rabbit IgG (diluted 1:100) or with FITC-labeled goat anti-mouse IgG Ab (diluted 1:500). Cells were washed again and re-suspended in 500 μL PBS. Ab binding was measured by flow cytometry using a FACSCalibur (BD Biosciences). FITC and PE dyes were excited by an argon ion laser at 488nm, which yields emission of 519nm and 575nm, for these two dyes, respectively. The data were analyzed by using the WinMdi or cyflogic software (BD Biosciences).
Expressing polyclonal ScFv antibodies
For the expression of the selected soluble ScFv proteins, the E. coli nonsuppressive bacterial strain, HB2151, was transfected with phagemid vectors. The bacteria were grown in a 2xTY medium containing 0.1% glucose and 100 μg/mL ampicillin, until the optical density at 600nm reached ∼0.8–0.9. Induction was performed with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) for a 3 h growth at 30°C. Peri-plasmic extracts, containing the ScFvs, were prepared by extracting the pelleted bacterial with 20% sucrose and 1 mM EDTA (pH 8.0) on ice. The ScFv Abs-containing fractions were collected after centrifugation at 10,800 g for 15 min at 4°C.
Expression of monoclonal ScFv antibodies for screening
Expression of ScFv mAb was performed by using 96-well bacterial plates (from Nunc) containing 2xTY medium containing 1% glucose and 100 μg/mL ampicillin. Single colonies of HB2151 bacteria, transfected with phagemid vectors from either the third or the fourth selection, were chosen for each well. After an overnight growth at 37°C, the bacteria from each well were diluted 1:50 in a new plate well with fresh 2xTY medium containing 0.1% glucose. Glycerol (to a final of 15% V/V concentration) was added to the original plates and they were stored at −80°C. The diluted cultures were grown for 3 h at 37°C, and then induced overnight with 1 mM IPTG at 30°C. After spinning the plates at 3500 g for 30 min at 4°C, the soluble ScFvs-containing supernatants were collected for further studies.
ELISA screening of the monoclonal ScFv Abs (with cells expressing CCR5)
All procedures were performed at room temperature. 3T3.T4 or 3T3.T4.CCR5 cells were grown in 96-well plates (∼10,000 cells/well) for 15 h. After centrifuging the plates at 210 g for 6 min, the media were removed and the cells were blocked for 1 h with PBS, supplemented by 2% (w/v) FCS and 2% BSA (FPBS). Each incubation was followed by a similar centrifugation of the plates at 210 g for 6 min, and then re-suspending the pelleted cells. Then, the cells were incubated for 1.5 h with the monoclonal Ab supernatants, diluted in FPBS, and then rinsed extensively with PBS. At this stage, the cells were incubated for 1 h with anti-c-Myc mAb (diluted 1:100 in FPBS) following by washings three times in PBS. The last incubation of the centrifuged cells was performed with a secondary Ab, anti-mouse IgG conjugated with HRP at a 1:1000 dilution. Color was developed after adding 100 μL 2,2-azido-bis (3-6 sulfonic acid) (ABTS) and the extent of binding was measured by the absorbance at 405nm.
IgG expression and purification in E. coli
The heavy and light IgG chains were expressed in E. coli BL21(DE3) pUBS500 cells that were transfected with either pHAK-IgH or pHAK-IgL plasmids with 100 μg/mL ampicillin and 50 μg/mL kanamycin at 37°C. The bacterial cultures were induced for protein expression at the late log growth phase (OD600nm of 2.5–3.0) with 1 mM IPTG for 3 h at 37°C. The recombinant protein accumulated as insoluble inclusion bodies that were isolated from the lysed bacteria by centrifugation. From 500 mL of culture, ∼2 g of wet cell paste was collected. The cells were suspended by sonication in 70 mL of TE (50 mM Tris-HCl and 20 mM EDTA, pH 8.0) and lysed by gentle rotation in 40 mg of lysozyme at 25°C for 1 h. The cell lysate was adjusted to 300 mM NaCl and 1.5% (v/v) Triton X-100 and disrupted using by sonication. The insoluble fraction was collected by centrifugation at 10,000 rpm in a GSA rotor (Sorvall) for 50 min at 4°C. This crude inclusion bodies preparation was further purified by two additional cycles of sonication in the same buffer (with 1% v/v Triton X-100) followed by centrifugation at 10,000 rpm in a GSA rotor for 30 min at 4°C. Finally, the inclusion bodies were treated again in the same way, except that the buffer contained no detergent. The inclusion bodies were completely solubilized in TE containing 6 M guanidine hydrochloride, incubated at gently rotation for 2 h at 25°C; the supernatant was collected after centrifugation at 10,000 rpm for 30 min at 4°C. The inclusion bodies were mixed at a heavy-light chain molar ratio of 1:2. The inclusion bodies mix was then reduced with 10 mg/mL DTE for 2 h at gentle rotation at 25°C. The solubilized reduced protein were refolded by X100 fold dilution into inclusion bodies refolding solution, containing redox shuffling and aggregation-preventing additives (oxidized and reduced glutathione, and L-arginine) (48), for 36 h at 8°C. After refolding, the protein was dialyzed against phosphate buffer pH 7.4. The refolded active protein was then filter sterilized using 0.45 μm filter and finally separated from the bacterial proteins, excess light chains, and improperly folded proteins by protein-A Sepharose chromatography.
Production of the 293-RED-CCR5 cell line
Human CCR5 cDNA was amplified by PCR reaction. The resulting 1.1 kb PCR product was cloned into the mammalian expression vector, pDiHcRED (20) containing red fluorescent reporter gene. CCR5-expressing cell line was generated by transfection of 293T cell line with the pDiHcRED-CCR5 expression vector.
Recognition of the recombinant A2-IgG by peptides representing the ECLs of CCR5
This ELISA-based procedure was performed at room temperature. Microtiter ELISA plates (Nunc) were coated overnight with NeutrAvidin (2 μg/well in 100 μL). The coated wells were subsequently blocked for 1 h with PBS, supplemented by 2% (w/v) skimmed milk powder (MPBS). Then, the wells were incubated for 1 h with biotin-conjugated peptides, designed by us and representing the first and second ECL of the CCR5 protein (Fig. 8) diluted in MPBS. After extensively rinsing with PBS containing 0.05% (V/V) Tween-20 (TPBS) and with PBS, the wells were incubated for another 1 h with the A2 IgG or the control–2D7 Ab and washed three times in PBS. The last incubation performed with a secondary Ab; HRP conjugated anti-human IgG (for the A2 IgG), and anti-mouse IgG (for 2D7 Ab) at 1:1000 dilutions. Color was developed with 100 μL ABTS solution and the extent of binding was measured by the absorbance at 405nm.
Quantification of HIV-1 p24
HIV-1 p24 antigen-capture assay kit (from the AIDS Vaccine Program, Frederick, MD) was used to determine the amount of p24 in the culture medium, in accordance with the standards and instructions supplied by the manufacturers. All work with infectious HIV-1 was performed in a P3 laboratory.
Protein analysis by polyacrylamide gel electrophoresis (PAGE)
SDS-PAGE was performed, under either reducing or nonreducing conditions, as described in detail elsewhere (52,60).
Western blot analysis
The protein bands, resolved by SDS-PAGE, were electro-transferred onto a nitrocellulose membrane. The membrane was blocked for at least 1 h with PBS supplemented by 5% (W/V) non-fat milk powder at room temperature with slow agitation. The membrane was then washed several times with PBS, followed by incubation with the specific primary mouse anti c-Myc or rabbit anti human IgG, followed by appropriate HRP-conjugated secondary antibodies. After three washes with TPBS and one wash with PBS, the nitrocellulose filter was developed with the Thermo Scientific Pierce ECL Western Blotting Substrate as described by the supplier. The membrane was then exposed to an X-ray film.
Results
Generating 3T3.T4.CCR5 cells that express high GFP levels
In order to screen the human antibody phage display library for phages, which specifically bind different domains on the cell-surface expressed CCR5, we have used a mixture of two cell types: live cells that over-express on their surface the target protein and excess control cells not expressing CCR5, which are supposed to reduce nonspecific background binding. We further co-expressed the marker GFP protein in the CCR5-expressing cells. The gfp gene was transduced into the 3T3.T4.CCR5 cells by a lentiviral-mediated gene transfer, yielding 2.9% high GFP-expressing cells (Fig. 1, A1). The population of the sorted GFP-expressing cells was further sorted twice, thus generating a homogenous cell population that expresses high levels of both GFP and CCR5. A flow cytometry analysis showed that 20.5% of the cells expressed high levels of both CCR5 and GFP (Fig. 1, A2). These cells were further sorted, so that 39.7% of the cells express high GFP and CCR5 levels (Fig. 1, A3). A flow cytometry analysis of the finally-selected positive cells (designated 3T3.T4.CCR5-GFP) shows that 96.3% expressed the GFP marker (Fig. 1B).
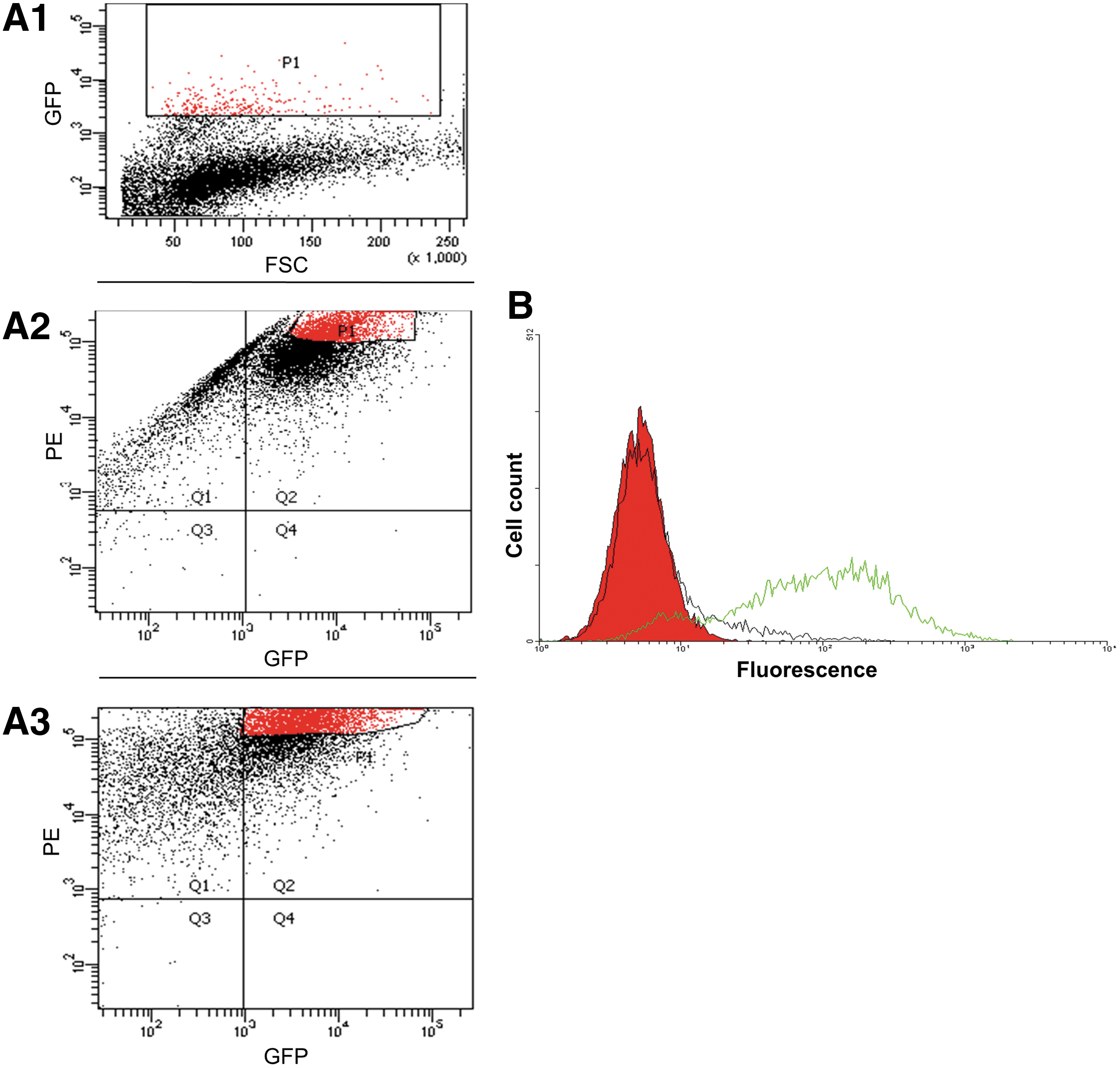
Selection for the 3T3.T4.CCR5-GFP cells that express high levels of both GFP and CCR5.
Affinity selection, screening, and enrichment of phage display libraries against CCR5-expressing cells
A mixture of two phage display libraries, Metha1 and Metha2 (44), each displaying human ScFv Abs, was used for isolating specific anti-CCR5 Abs. Altogether, four selection cycles were performed. Since each specific antibody is present at low quantities in the original libraries, sorting was done only from the second selection cycle to avoid losing any specific binders at this step. At all selection stages, 1×1012 pfu of phages were added. The first step was performed as follows: after blocking with BSA, the initial libraries were mixed with ∼2×107 3T3.T4.CCR5-GFP cells. After extensive washings, the cells were lysed and the whole solution with infectious phages was used for amplification and further selection cycles, performed each with a mixture of 4:1 ratio of the control cells 3T3.T4 over the 3T3.T4.CCR5-GFP cells (a total of 8×107 cells). This cell mixture step is critical for the employed technique, since by incubating the phages with an excess of control cells, which express all cell surface epitopes except for CCR5 (as well as GFP), most nonspecific phages that bind unrelated proteins interact with the control cells and, therefore, are not expected to be selected during sorting. To enrich for the desired phages, the libraries were screened with these cell mixtures in three consecutive selection cycles. A schematic description of the sorting-selection procedure is shown in Figure 2. After each selection step, phages were eluted from the cells by cell lysis and then used to infect susceptible E. coli TG1 bacteria. After four selection rounds, polyclonal phage populations from each selection step were tested for binding either 3T3.T4 (negative) or 3T3.T4.CCR5 (positive) cells using flow cytometry (after staining the phages with anti-phage fd protein specific Abs). Since no GFP was expressed at this stage by the CCR5-expressing cells, the only apparent difference between the positive and negative cells is the over-expression of CCR5. Therefore, it is likely that the substantial selective binding of the phages to the 3T3.T4.CCR5 cells is primarily due to their interaction with CCR5, thus leading to phage enrichments (Fig. 3A).

A schematic description for selecting the phage display libraries. Phages were incubated with 3T3.T4.CCR5-GFP cells along with an excess of the depleting nonlabeled 3T3.T4 cells. After phage binding, the cells were sorted and collected by flow cytometry according to their GFP fluorescence. These cells are enriched in the specific population of the phages that bind specifically the CCR5 protein (marked by brown dots) relative to the nonspecific binders (marked by black dots). Color images available online at
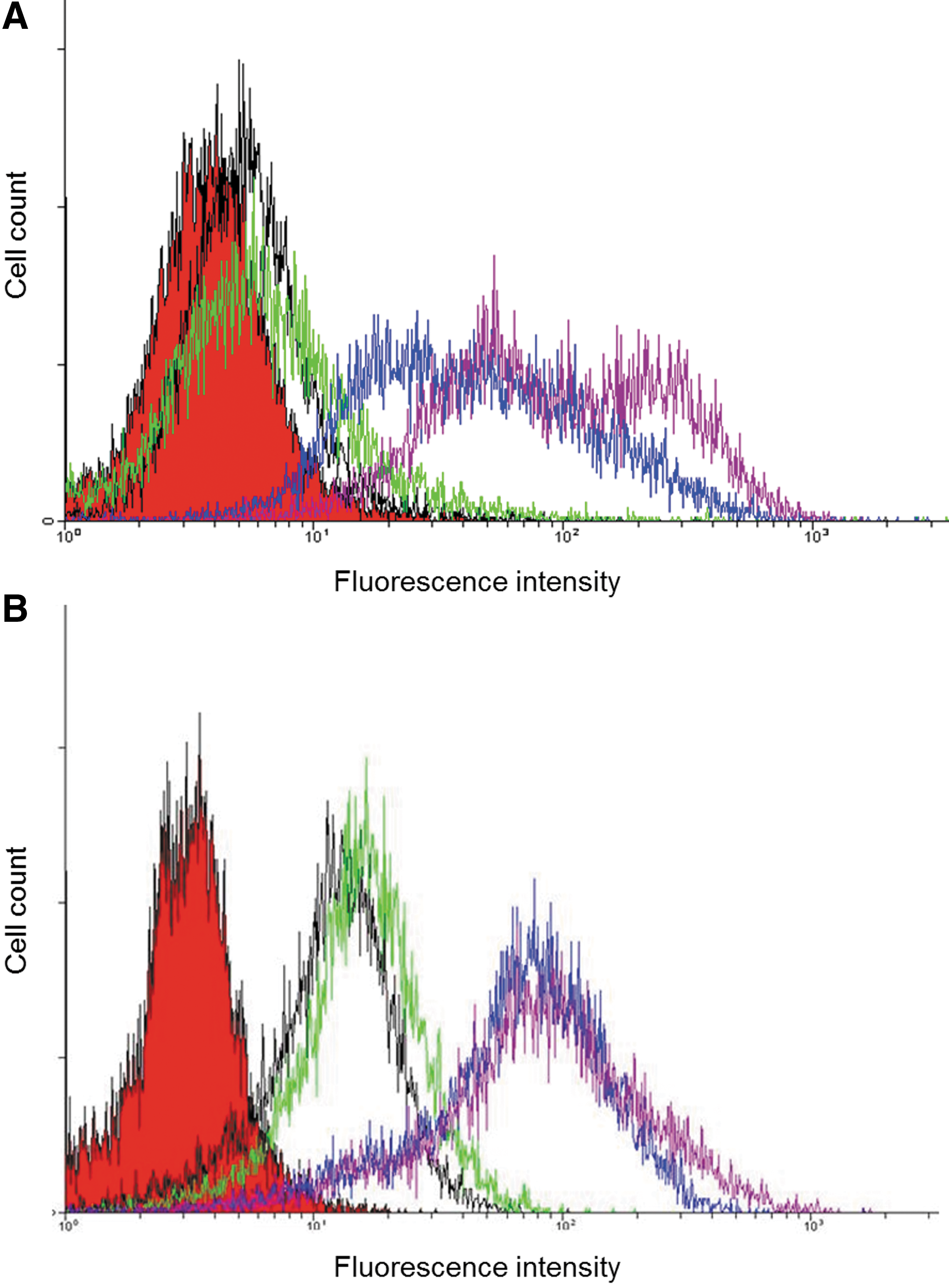
Flow cytometry analysis for polyclonal phage and Abs populations obtained after four selection cycles.
The polyclonal ScFv Abs selected against CCR5
The next step involved the isolation of soluble ScFv that were expressed by the selected polyclonal phage population. To this aim, we tested the ScFvs expressed by the phages, selected in the third and fourth cell sorting cycles. Nonsuppressor E. coli HB2151 cells were infected with the phages, present in the cell lysates, and the bacteria were induced to express soluble ScFvs by IPTG. The bacterial peri-plasmic extracts, containing the ScFvs were tested. The peri-plasmic fractions, which were prepared from each selected phage population, contained the ScFvs that could be detected by anti-c-Myc mAb (as c-Myc epitope is part of the C-terminus of all ScFvs, data not shown) in a Western blot. Similar amounts of soluble mAbs were detected in all fractions. Then, the ScFv Abs-containing fractions were tested by flow cytometry analysis for recognizing cells expressing surface CCR5. Indeed, these Abs that were derived from the third and fourth selection cycles showed a substantial enrichment for specifically-interacting with CCR5-expressing cells (Fig. 3B).
Selecting for ScFv mAbs specific to the cell-surface expressed CCR5
The phage populations from the third and fourth panning cycles were used as a source for testing the ScFv mAbs. These soluble mAbs, expressed by the HB2151 bacteria, were tested by ELISA for binding CCR5 on the CCR5-expressing cells. Out of a total of 474 screened individual clones, five were found to bind the CCR5-expressing cells selectively (all derived from the fourth selection cycle). An ELISA analysis of these clones is shown in Figure 4A. These five selected anti-CCR5 clones were further analyzed by flow cytometry, using the bacterial peri-plasmic fractions that contained the specific soluble ScFvs. Clones A2, H7, and M2 exhibited a strong specific binding to cell expressing CCR5, albeit to a different degree (Fig. 4B). Out of them, A2 showed a higher binding capacity to the CCR5-expressing cells.
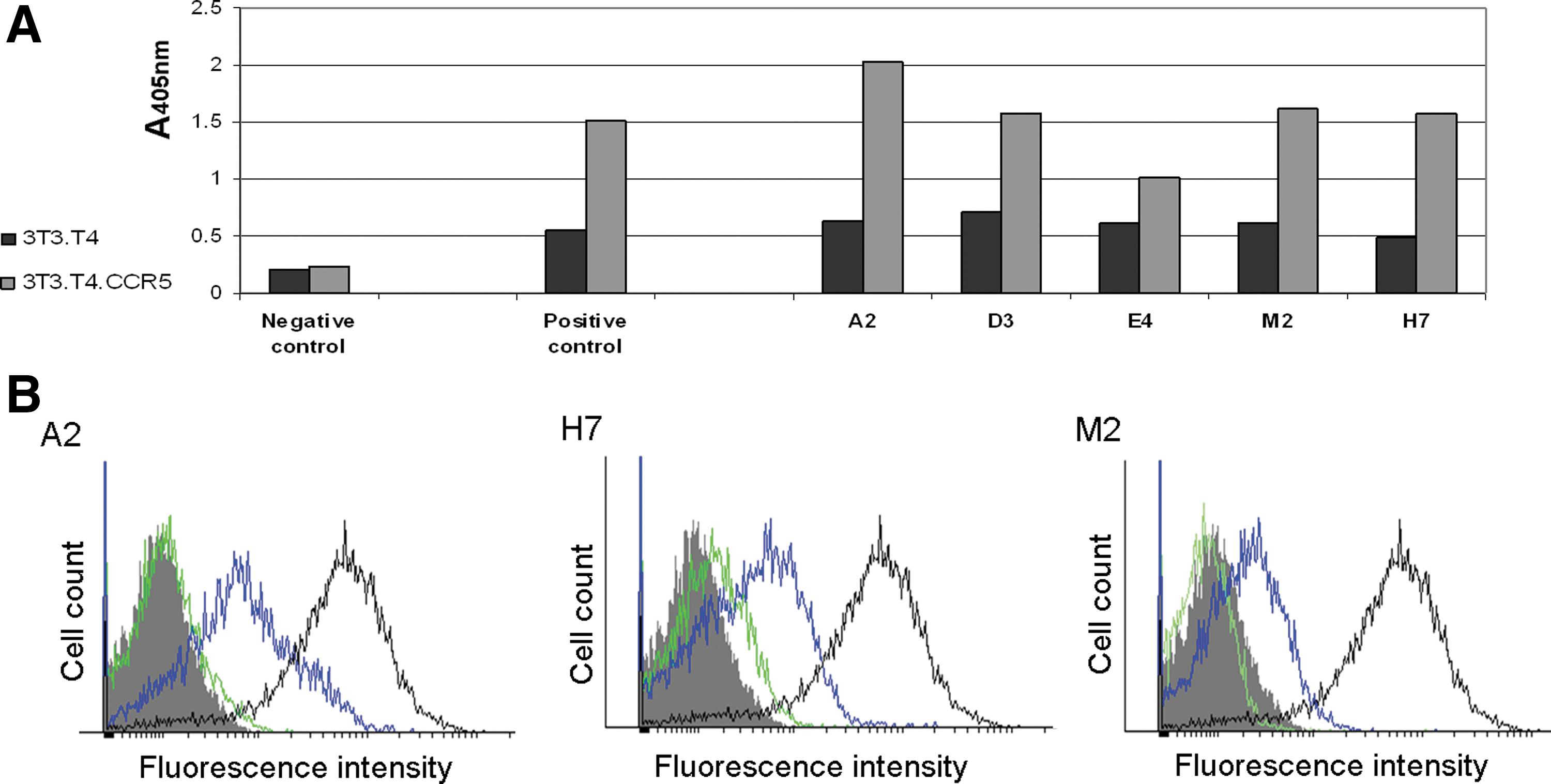
ScFv mAbs.
Sequence analyses of selected ScFv mAbs
To test the novelty of the anti-CCR5 mAbs isolated in this study, we have sequenced the genes encoding the five ScFv mAbs with the strongest binding to CCR5 (i. e., those designated A2, M2, H7, E4, and D3). The sequences were compared to known published Abs genes, using Igblast (

The amino acid sequences of the five selected anti CCR5 ScFvs mAbs. Amino acid sequences of the mAbs, designated A2, M2, H7, E4, and D3, were designated with one-letter codes and aligned employing the EMBOSS Pairwise Alignment Algorithms (
Preparing and testing the A2 IgG mAb
In an attempt to improve binding ability of A2, the ScFv with the highest affinity towards CCR5, A2-ScFv, was converted into a full-length human IgG format, with two identical Ag binding sites for its target. The A2 ScFv-encoding gene was used as a template for constructing polymerase chain reaction (PCR) products that allow expressing the L and H chains in pHAK expressing vectors. This resulted in high expression level in bacterial cells of the A2-IgG in L and H chains (see Materials and Methods) (22). The purified IgG Ab was analyzed by 10% SDS/PAGE under either nonreducing (data not shown) or reducing conditions (Fig. 6A). The A2-IgG was further studied by flow cytometry over a wide concentration range. Specific dose-dependent binding could be detected from as low as ∼4 nM and minor nonspecific binding to control cells was detected at only high concentrations (Fig. 6B and C).
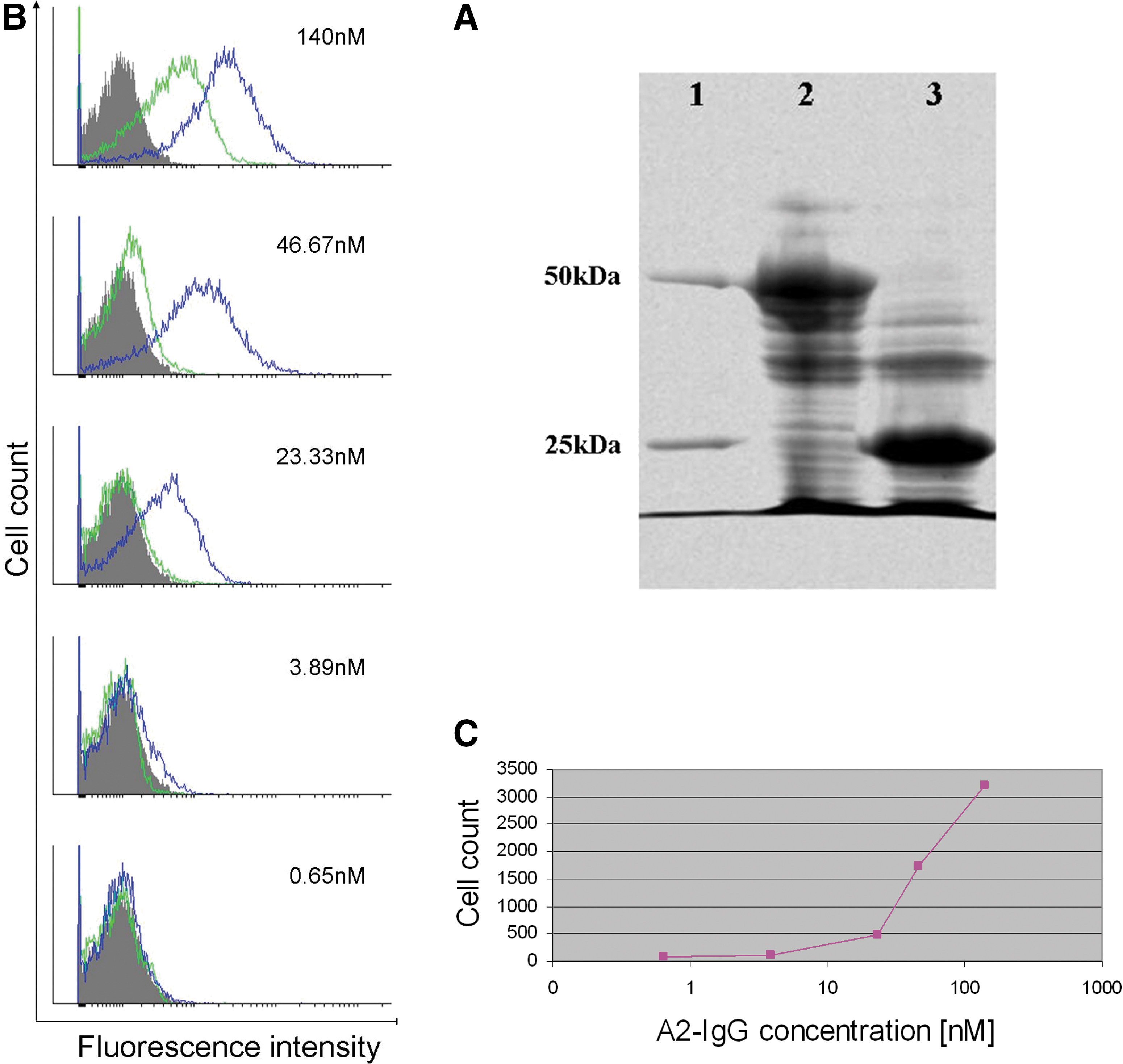
Analysis of the A2-IgG.
Next, we verified the specific binding of the A2 mAb to the CCR5 co-receptor. For this purpose we have generated cells expressing both RED fluorescent protein and CCR5, by producing a fused protein of both, designated 293T-DiHcRED-CCR5. These cells were tested for the present of CCR5, by using confocal microscopy after incubating the cells with the commercially-available anti-CCR5 (2D7-mouse anti-CD195 mAb), followed by a secondary rabbit-anti mouse FITC-labeled Ab. The results confirm the specific binding of 2D7 to the co-receptor (Fig. 7A). Incubation of A2-IgG along with the 293T-DiHcRED-CCR5 cells showed a specific binding to CCR5 (Fig. 7B). Images of the binding were used to analyze the co-localization of A2-IgG and CCR5. A program written in MATLAB (a high-level technical computing language for algorithm development, data visualization, data analysis, and numerical computation) decomposed the color images into their RGB (red, green, blue) components. The relative contents of green color at each pixel of the image was calculate for each image (NG=G/(R+G). The co-localization image NG was constructed in a jet color map format. The average co-localization was calculated, and it was found that 85.7% of the red fluorescent protein-labeled CCR5 was also stained with the green fluorescent-labeled A2-IgG.
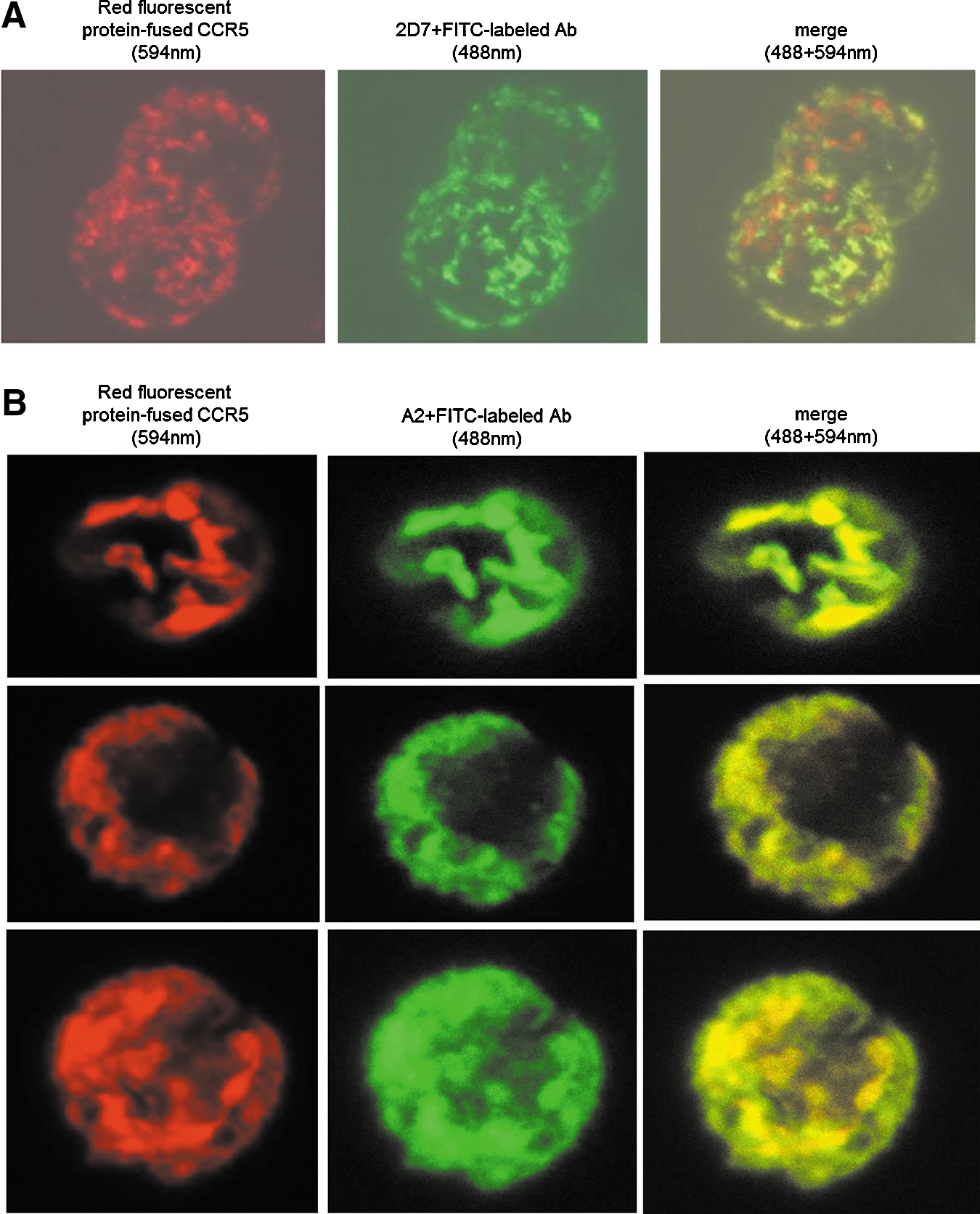
Verification of A2 mAb specificity towards CCR5, using confocal microscopy.
Testing A2 IgG against the first and second extracellular loops of CCR5
To test which specific CCR5 domains interact with the newly generated A2- mAb IgG, we used three synthetic peptides derived the first and second ECLs of CCR5. This examination was done in comparison with the mouse anti-CCR5 IgG mAb-2D7. Peptide No. 1 was derived from the first ECL of the CCR5 co-receptor, while the peptides, designated 2a and 2b, are the linear and cyclic peptide, respectively, both derived from the second ECL of CCR5 (Fig. 8). Epitope mapping of A2 IgG Ab was done by ELISA using these peptides and suggested that the second ECL of CCR5 (as either a linear or a cyclic sequence as the epitope) interact with A2- mAb (Fig. 9). There is, however, a marginal preference towards the linear peptide. Interestingly, the second ECL of CCR5 was previously believed to contain the dominant epitopes for highly potent anti-HIV mAbs (62).
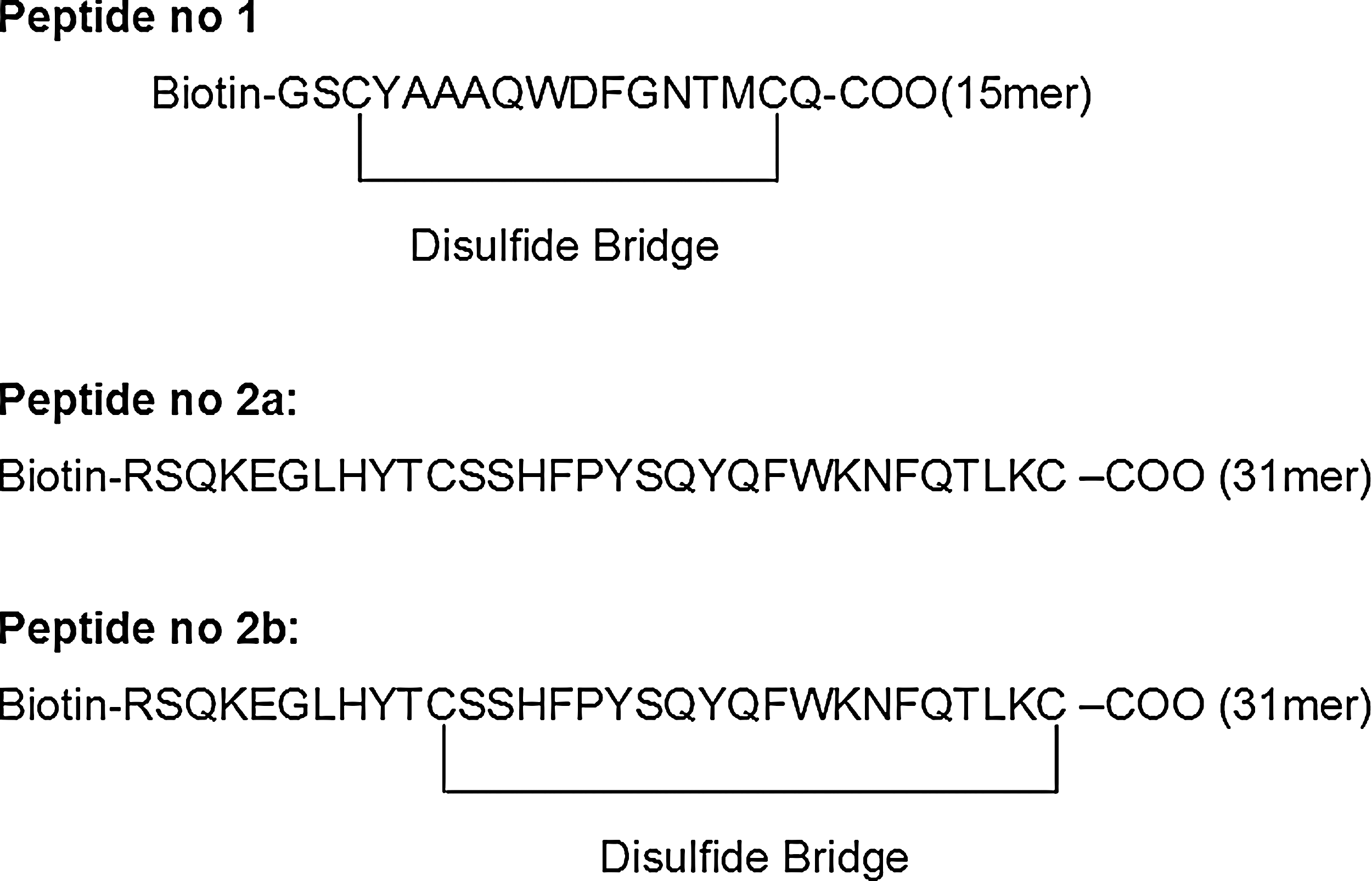
Design of N-terminal biotinylated peptides derived from the CCR5 epitopes. Three peptides were designed by us and were synthesized by GL Biochem, China. Peptide No. 1 was derived from the first ECL of the CCR5 co-receptor (from Tyr-89 to Gln-102), while peptides No. 2a and 2b were derived the second extracellular loop of the CCR5 co-receptor (from Arg-168 to Lys197). Peptides 2a and 2b have the same primary sequence, but were synthesized as the linear and cyclic forms, respectively.
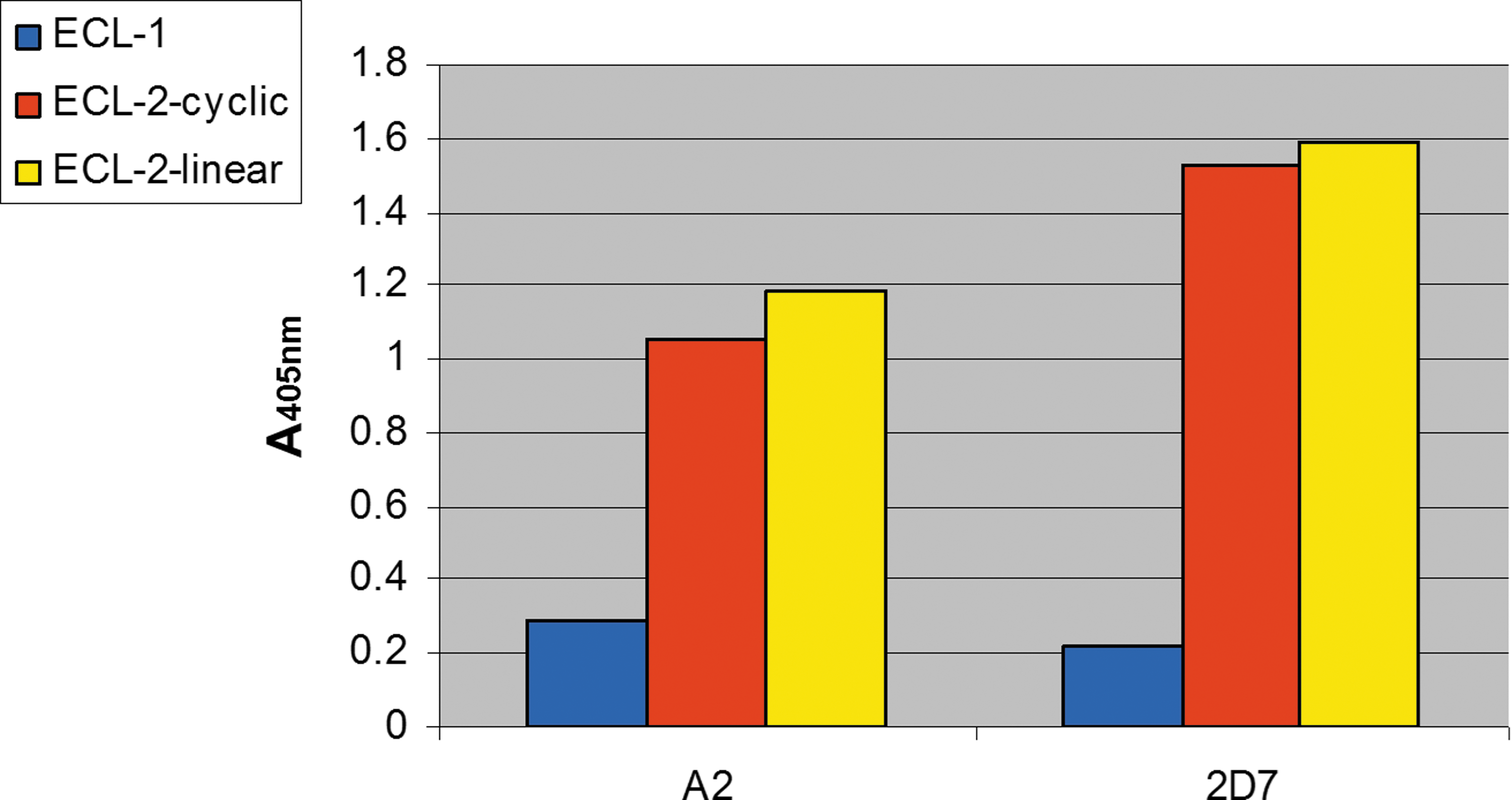
Binding of the human A2-IgG (in comparison with the mouse 2D7-IgG) to the studied CCR5 ECL2-related peptides. This ELISA-based assay was performed as described in Materials and Methods with the peptides described in Figure 8. Color images available online at
Discussion
The purpose of this study was to develop a relatively simple and efficient technique for screening phage libraries that present human Abs, in order to identify mAbs that specifically interact with cell surface proteins. As a test case to accomplish this goal, we decided to identify novel anti-CCR5 mAbs and, hence to assess the potency of these Abs. There is evidence that the binding of mAbs to different epitopes of CCR5 on the cell surface, can result in different inhibitory effects (2,50). Since CCR5 is the major coreceptor for HIV-1, the new CCR5-neutralizing Abs are supposed to target HIV-1 sensitive cells and, consequently, may have anti-HIV activities. It is well accepted that the presence of CCR5 is not critical to cell viability, since large deletions in this protein do not seem to affect the expressing cells (29). Therefore, the potential advantage of blocking CCR5 is that it should not be detrimental to normal cells, thus minimizing the side effects. Consequently, several CCR5 mAbs were already isolated and characterized and some of them demonstrated potent anti-HIV activities (7,36,42,54,57,59,61). However, the lack of specificity of several available anti-CCR5 mAbs may impede their use (6). Therefore, further studies are required to identify additional highly-specific and more efficient mAbs that can block viral entry.
In the hereby presented technique, we screened a mixture of two phage display libraries, expressing human ScFv mAbs, against cells that present on their surface CCR5. The main uniqueness of this method relies on the use of an excess of control cells that do not express the CCR5 molecule (and the marker GFP) and are otherwise identical to the CCR5-positive cells. The use of an excess of control cells allowed exclusion of phages that bind nonspecifically to these cells. Thus, most phages that express human ScFv mAbs and bind the extracellular domains of native CCR5 will be eventually selected. Quantitative cell sorting allowed the separation of the CCR5/GFP positive cells, to which the specific phages were bound. After phage elution, the selected phages were re-screened by further affinity selection rounds. Phage populations obtained after the third and fourth panning were used as a source for screening for soluble ScFv mAbs expressed by bacteria. Out of individual clones that were evaluated by ELISA, five were found to bind the CCR5-expressing cells selectively. The clones, designated A2, H7, and M2, were found to exhibit the highest specificity towards the CCR5 co-receptor. As seen in Figure 5, all three ScFvs are composed of unique sequences.
The ScFv Ab designated A2, with the highest affinity towards CCR5, was produced as an IgG1 format (22). Using synthetic peptides, derived from CCR5 ECLs, we analyzed which specific CCR5 domains interact with the A2-IgG mAb. The data show that the epitope recognized by the A2-IgG Ab is contained within the second ECL of CCR5 (although its binding was weaker than that of the commercial mouse 2D7 Ab). This specific ECL is believed to contain the dominant epitopes for highly potent anti-HIV-1 mAbs (38,62). New studies support the view that the most potent anti-CCR5 mAbs recognize the second CCR5 ECL, either exclusively or in combination with the protein's N-terminus (30,31). Studies have revealed fundamental differences in the way mAbs and small molecules bind CCR5 and inhibit HIV-1. CCR5 mAbs and small-molecule CCR5 antagonists were shown to have a consistent antiviral synergy (30,31). Interestingly, single intravenous infusions of CCR5 mAbs significantly reduced HIV-1 RNA levels in HIV-1 infected individuals for 2–3 weeks without appreciable toxicity (51). However, it should be emphasized that a CCR5 binding activity does not necessarily imply an anti-HIV-1 activity, as the recognition of surface CCR5 varies depending on the cell type used. Therefore, one cannot assume that each CCR5-binding Ab will also recognize this epitope on the surface of all primary CCR5-expresiing cells.
It should be also mentioned that ScFv M2, which showed a relatively high affinity towards CCR5 (Fig. 4) and had a sequence that is different from A2 (Fig. 5), was converted also to the IgG format. Like the A2 IgG, the resulting M2 mAb IgG also bound the second ECL of CCR5 (data not shown). However, although the M2 mAb IgG bound with high affinity to the Cf2Th/syn CCR5 cell line, it failed to show significant binding to the 3T3.T4.CCR5 cells (data not shown). This suggests that not every epitope-interacting antibody recognizes every cell that presents this epitope as part of the intact receptor. In this specific case, it is likely that the low expression of the CCR5 protein on the 3T3.T4.CCR5 cells (relative to the Cf2Th/syn CCR5 cells) (47) was below the level of detection by M2 IgG. Therefore, this Ab was not further studied by us.
The isolated ScFv-mAbs can be further explored for therapeutic application in HIV therapy by blocking the virus interaction with its co-receptor. These novel inhibitors can undergo modifications in order to improve their potency, stability, and bioavailability properties. Such improvements can include affinity maturation (of A2 CDR3) for producing modified A2 Ab versions with increased affinity towards the CCR5 co-receptor (56) or chemical modification of the mAbs that may induce the internalization of the CCR5–Ab complex. For example, the addition of amino-oxypentane at the N-terminus has been reported to inhibit the recycling of the bound receptor that otherwise may reappear on the cell surface after ligand removal (41). The described methods can serve also as a lead for isolating mAbs that induce internalization of the co-receptor, by additional phage selections that will be performed at 37°C with trypsinization to remove extracellular-bound phages. Under these conditions, phages that are not internalized will be removed; thus only phages that were internalized (and hence located inside the GPCR-expressing cells) will be selected (5).
In summary, the presented results show that the developed screening procedure is indeed applicable for selecting the appropriate Abs. Moreover, it is important to note that this new approach can be also applicable for screening, isolation, and enrichment of phage display libraries against practically every cell protein with extracellular domains, such as other GPCRs, transporters ion channels, and receptors of every virus bacteria or any other pathogens (all difficult to obtain as native purified proteins). Hopefully, the newly generated ScFvs and A2-IgG versions will open innovative opportunities for therapeutic applications and for the development of novel anti-HIV/AIDS drugs.
Footnotes
Acknowledgments
We wish to thank Dr. W. Marasco, Dana Farber Cancer Institute, Harvard Medical School, Boston, Massachusetts, for kindly providing the Metha 1 and Metha 2 phage display libraries. In addition, we would like to thank all the following people from Tel Aviv University, Dr. E. Bacharach for supplying the HIV PV system, Dr. Y. Oshri for his help with the flow cytometric experiments, Dr. K. Hirschberg for help in preparing ![]() , and to Dr. I. Oz-Gleenberg (from our laboratory at Tel Aviv University) for critically reading the manuscript, Dr. Noel Exelrod for his help with MATLAB analysis. We are also grateful to the AIDS Research and Reference Reagent Program for supplying the following cell lines: 3T3.T4, 3T3.T4.CCR5, Cf2Th-CXCR4, and Cf2Th/syn CCR5.
, and to Dr. I. Oz-Gleenberg (from our laboratory at Tel Aviv University) for critically reading the manuscript, Dr. Noel Exelrod for his help with MATLAB analysis. We are also grateful to the AIDS Research and Reference Reagent Program for supplying the following cell lines: 3T3.T4, 3T3.T4.CCR5, Cf2Th-CXCR4, and Cf2Th/syn CCR5.
This work was performed in partial fulfillment of the requirements for a Ph.D. degree of M. Shimoni at the Sackler School of Medicine of Tel Aviv University.
Author Disclosure Statement
All authors declare that they have no competing financial interests.