
Abstract
The present study describes the strains of hepatitis C virus (HCV) isolated from Tunisian hemodialysis patients. Thirty-three HCV strains isolated from different dialysis centers in Tunis City were amplified by RT-PCR in a region of the NS5b gene, genotyped by sequencing, and compared to international sequences by phylogenetic analysis. The phylogenetic tree showed that 16 HCV isolates have been identified as subtype 4k (48.5%), 7 as unspecified HCV-4 subtype (21.2%), 5 as subtype 4a et 1b (each 15.2%). The analysis of this tree revealed that the HCV-1b strains were closely related to Anglo-Saxon and European isolates, while the HCV-4 isolates are genetically similar to Egyptian and African strains. Phylogenic analysis of 33 Tunisian isolates with international HCV strains on a region of the NS5b gene demonstrated that the subtype 4k submerged the Tunis city and a new subtype of HCV4 seems to be suspect in this area.
Introduction
HCV is an enveloped single-stranded, positive-sense RNA virus. It belongs to the flaviviridae family. The genome of this virus is characterized by a high degree of variability due to the poor fidelity to the viral RNA-dependent RNA polymerase and the lack of genome repair mechanisms (2). Analysis of the NS5b region encoding the viral RNA polymerase from a wide range of HCV isolates led to the classification of HCV into six major genotypes and more than 70 subtypes (6,40).
Several epidemiological studies have shown that the HCV genotypes have a particular geographic distribution throughout the world and certain genotypes predominate in certain areas. Genotypes 1, 2, and 3 are distributed almost worldwide (25). Genotype 4 has been reported to be the most prevalent genotype in northern and central Africa, Middle East, and Egypt (7,19,37). Genotype 5 was found in South Africa and genotype 6 was especially located in South East Asia (32). In Tunisia, a sero-molecular-epidemiologic national inquest of HCV infection from hemodialysis patients was done in 2002 in the Charles Nicolle Hospital laboratory (3). This study showed that genotype 1b is the most prevalent (70.8%), followed by genotype 4 (21.2%). The regional distribution of genotype 4 was variable with predilection in the Tunis region (18.8%). The phylogenetic analysis of HCV genotype 4 has not been treated previously in Tunisian studies. This work is a first report that has focused on phylogenetic analysis of hepatitis C virus genotype 4 isolates from hemodialysis patients in the Tunis city and this by comparing 33 isolates, which were selected from isolates of genotype HCV4 determined by Inno LiPA test during the national survey, with international HCV strains in a phylogenetic approach.
Materials and Methods
Patients and isolates
Thirty-three HCV strains were isolated from hemodialyzed patients in 21 dialysis centers in Tunis City (named A to X). The F unit is the only public center of the Charles Nicolle Hospital. It represents the basal center from which the majority of patients will be dispatched to their respective loco-regional private dialysis units. The number that follows the alphabetical letter indicates the patient number in the center. The 33 Tunisian HCV strains were labelled “T” (Tunisia), followed by a number (Table 1). Data obtained from each patient included age at diagnosis, gender, duration on hemodialysis, initial nephropathy, and possible risk factors for HCV (such as transfusion and/or surgery) were completed retrospectively at the hemodialysis centers (Table 2). All patients were negative for hepatitis B surface antigen (HBsAg) and human immunodeficiency virus (HIV). The HCV viral load was identified by HCV Real-TM Quant (Sacace, Biotechnologies) and revealed an average of 21489,667±42592,528 copies/mL in patients of this study. The use of recombinant erythropoietin to treat anemia due to renal failure was observed in one patient (S1). The notion of a stay abroad before dialysis treatment of end stage renal disease was identified in one patient (G1) in Saudi Arabia. None of the patients had received treatment for HCV infection before entering the study. In these hemodialysis units and during all the period of their dialysis, all the patients were tested for ALT levels (once/year). Thirty among 33 studied patients (91%) had normal levels of ALT (range: 5–55 U/L; mean: 46.89 U/L), and in 3/33 cases the cytolysis ranged from 1.5 to 3 times the normal of ALT with mean levels at 82.05 U/L.
n, number; N, normal; S, surgery; Tr, transfusion.
All patients had given informed consent, and the study was approved by the local ethics committee.
HCV typing with INNO-LiPA
The 33 Tunisian HCV isolates were typed by using the reverse hybridization line probe assay (INNO-LiPA assay; Immunogenetics, Belgium) according to the manufacturer's protocol.
RNA extraction and RT-PCR of NS5b region
RNA was extracted from patient serum by RNA columns (QIA amp viral RNA kit; QIAGEN) according to the manufacturer's instructions. Extracted RNA (12 μL) was transcribed for 90 min at 42°C using 0.2 mM of each dNTP, 20 pmol of random hexamers, 1x AMV buffer, 25 U RNasin, and 10 U of the RT AMV enzyme (Promega, USA) in a final volume of 25 μL.
The reaction volume of PCR was 50 μL containing 10 μL of cDNA, 0.2 mM of each dNTP, 1.5 mM of MgCl2, 0.5 U of Taq polymerase (Promega, USA) and 10 μM of each primer. Primer sequences (G4-980 and G4-981) (39) used are summarized in Table 3. The PCR programme is as follows: initial denaturation at 95°C for 15 min, 10 cycles of denaturation at 94°C for 40 sec, annealing at 50°C for 30 sec, and elongation at 72°C for 1 min; followed immediately by 35 cycles at 94°C for 30 sec, 50°C for 30 sec, and 72°C for 1 min per cycle. The final elongation step was at 72°C for 10 min. The PCR products (420 bp) were migrated on agarose gel (2%) and visualized under UV light.
Purification and sequencing
The amplified products were purified using QIAquick Gel extraction kit and were sequenced in the NS5b region of the viral genome by using ASN1121 primer (28).Cycle sequencing was undertaken using Cy5.0/Cy5.5 Dye Primer Cycle Sequencing, according to the manufacturer's instruction. The product was analyzed on an automatic TOWER MICROGene Clipper™ sequencer.
Phylogenetic analysis
Genetic similarities to the 33 HCV were identified with the BLAST program. Twenty-three international sequences from GenBank database (Table 4) were selected and aligned with the Tunisian isolates by using Clustal program W2.0 (23). Distances between sequences were determined with the DNA dist program (Kimura two-parameter model) of the PHYLIP package (20). The phylogenetic tree was constructed with the neighbour-joining algorithm (38) using the Mega 4 program (22).
AUS, Australia; BUR, Burundi; CAM, Cameroon; EGY, Egypt; FRA, France; GER, Germany; GRE, Greece; IRE, Ireland; SPA, Spain; TUN, Tunisia; UK, United Kingdom; USA, United States of America.
Nucleotide sequence accession number
The nucleotide sequences accession number of the 33 Tunisian strains and the 23 selected international HVC sequences are shown in Tables 1 and 4, respectively (1,13,26,32,33,38).
Results
HCV typing by INNO-LIPA
The 33 Tunisian HCV isolates were identified as HCV-4 by INNO-LiPA assay.
Epidemiological characteristics
As summarized in Tables 1 and 2, blood transfusion and/or surgery were identified as the most frequent risk factors in 19/33 (57.6%) of patients. Eight (24.2%) had no risk factor, and in 5 cases, the factors were unknown. Three patients with risk factors (R1, R2, and V2) were infected prior to 1995 during which the test screening for anti-HCV among blood donors was introduced in Tunisia country. In addition, the patient G1 had worked in Saudi Arabia prior to dialysis and was infected on admission by the strain T19 (HCV-4).
Phylogenetic analysis
The 33 isolates were previously typed by INNO-LiPA hybridization as HCV-4. The phylogenetic analysis of these strains performed in a partial region of the NS5b gene by comparing to the most genetically closed international sequences (23 sequences according to BLAST data) highlighted 5 clusters constituted of 4 subtypes (1b, 4a, 4k, 4o) and unspecified HCV-4 subtype (Fig. 1). Among the 33 Tunisian isolates, 16 were 4k (48.5%), 7 were unspecified HCV-4 (21.2%), and 5 were 4a and 1b (15.2% each). Among the 7 unspecified HCV-4 strains, one (T2) seems to be 4o subtype because it belongs to a cluster comprising exclusively HCV-4o Egyptian strains, while the 6 other unspecified HCV-4 strains (T3, T8, T15, T19, T36,and T37) cluster with 4o in a separate arm of the tree. In order to know the subtype of these isolates, a phylogenic analysis of these 7 unspecified HCV-4 isolates with 40 international HCV4 strains (Table 5), including all of the 19 assigned subtypes of genotype 4 in NS5b gene region, was done. The tree revealed that these isolates did not match any known subtype HCV4 but they are weakly related to isolates HCV4o (Fig. 2). Calculation of similarity between the HCV4 unspecified sequence and the 40 different sequences of HCV4 subtype showed similarity below to 90% (Table 6).
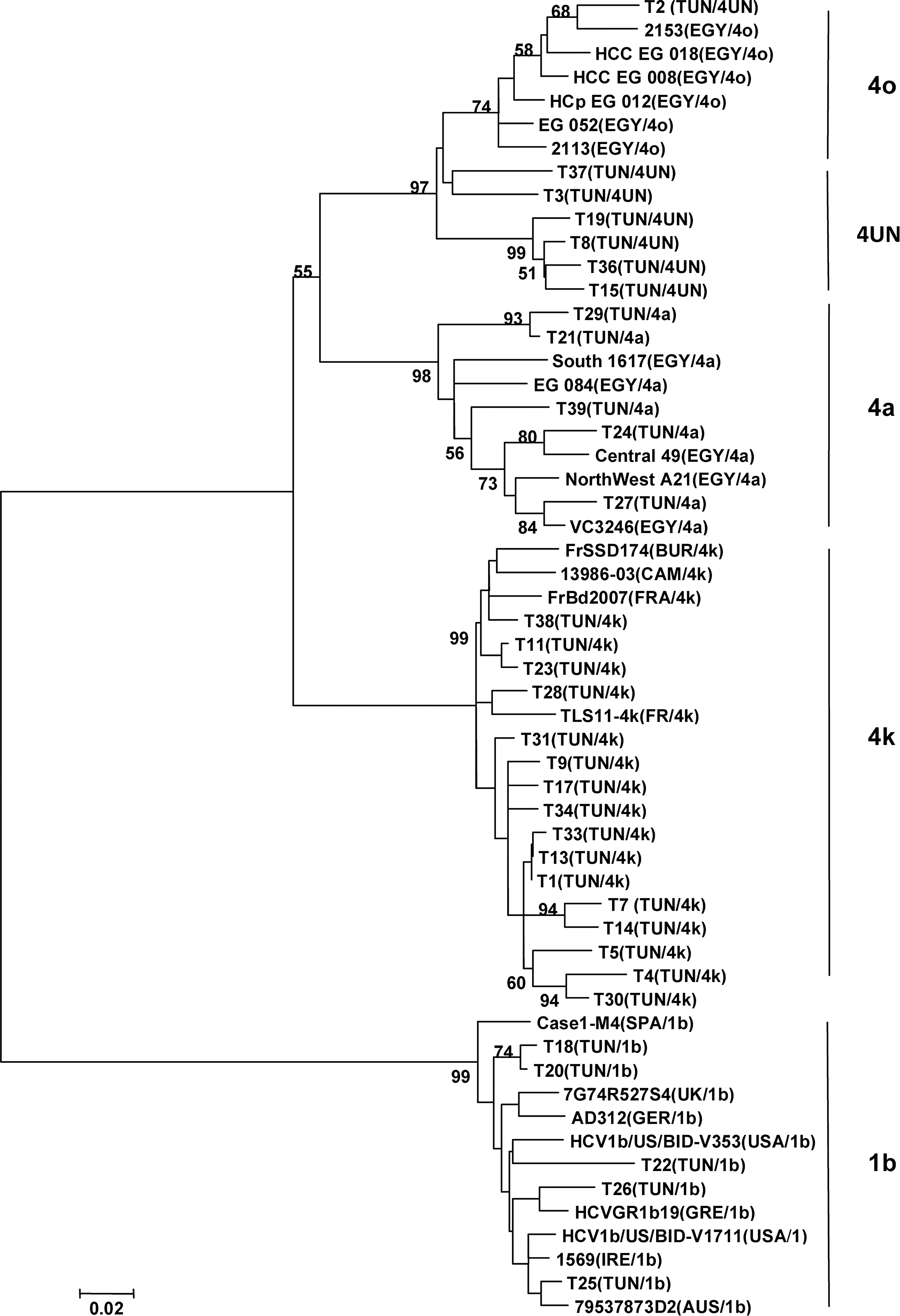
Phylogenetic tree of 33 Tunisian isolates and 23 international HCV strains on a region of the NS5b gene (nucleotides 8317 to 8581) designed by imputing the aligned sequences into the MEGA 4 program and constructed using the neighbour-joining algorithm. Genetic distances were calculated with the Kimura-2 parameter model with a transition/transversion ratio of 2.0, and the reliability of the tree was determined by bootstrap analysis with 1000 pseudo replicate data sets. For each strain, the county of isolation and the genotype/subtype are indicated in parenthesis. The bootstrap values are represented at the tree nodes, only those >50% are shown. The scale for the genetic distance is also represented. The countries are mentioned according to the following abbreviations: AUS, Australia; BUR, Burundi; CAM, Camaroon; EGY, Egypt; FRA, France; GER, Germany; GRE, Greece, IRE, Ireland; SPA, Spain; TUN, Tunisia; UK, United Kingdom; USA, United States of America.
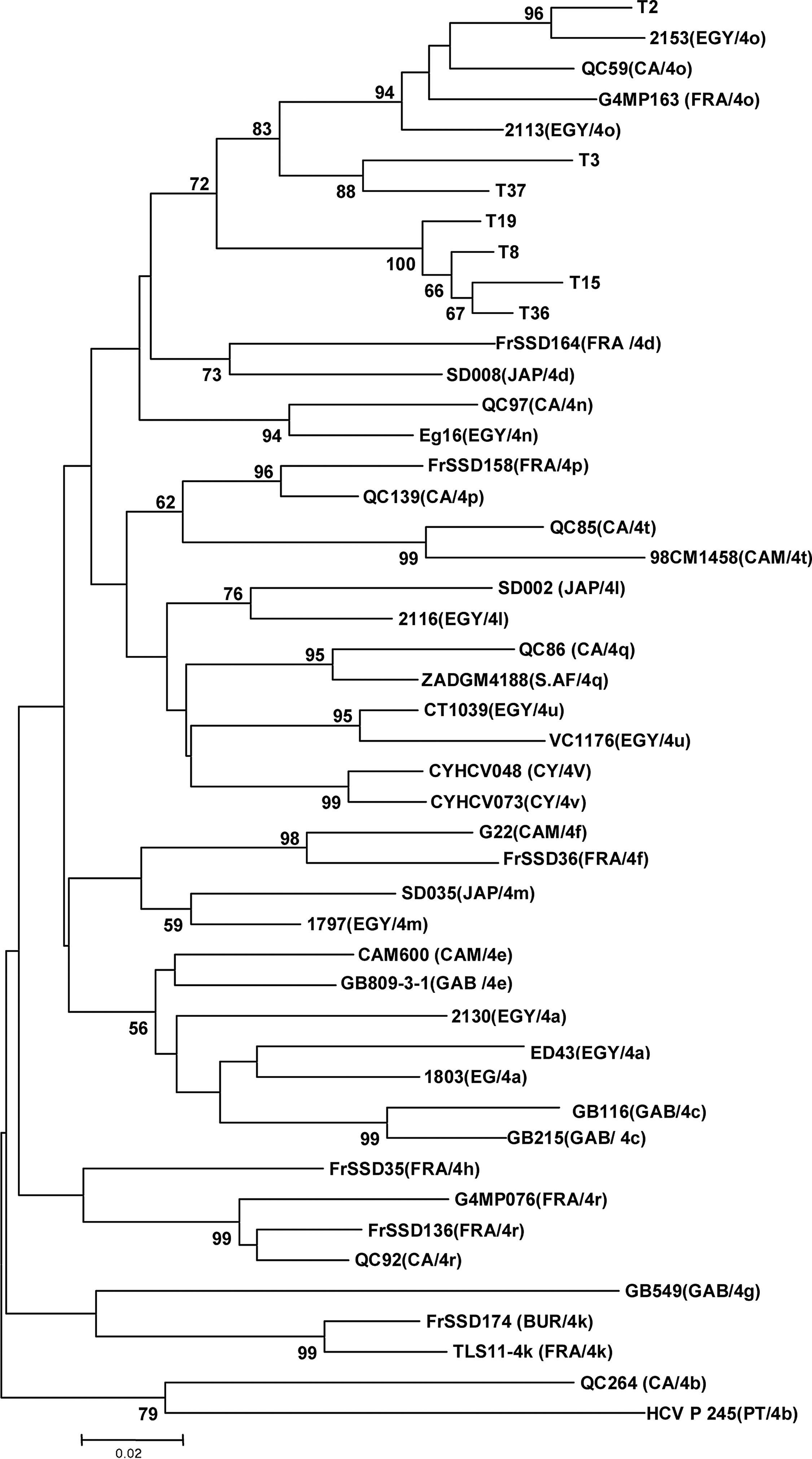
Phylogenetic tree of 7 unspecified HCV-4 isolates and 40 international HCV4 strains, including all assigned subtypes of genotype 4, on a region of the NS5b gene (nucleotides 8317 to 8581) designed by imputing the aligned sequences into the MEGA 4 program and constructed using the neighbor-joining algorithm. Genetic distances were calculated with the Kimura-2 parameter model with a transition/transversion ration of 2.0, and the reliability of the tree was determined by bootstrap analysis with 1000 pseudo replicate data sets. For each strain, the country of isolation and the genotype/subtype are indicated in parentheses. The bootstrap values are represented at the tree nodes, only those >50% are shown. The scale for the genetic distance is also represented. The countries are mentioned according to the following abbreviations: BUR, Burundi; CA, Canada; CAM, Camaroon; CY, Cyprus; EGY, Egypt; FRA, France; GAB, Gabon, JAP, Japan; PT, Portugal; S.AF, South Africa.
BUR, Burundi; CA, Canada; CAM, Cameroon; CY, Cyprus; EGY, Egypt; FRA, France; GAB, Gabon; JAP, Japan; PT, Portugal; S.AF, South Africa.
In the same way, the 5 HCV-4a Tunisian isolates are genetically close to Egyptian strains. On the HCV subtype 4k, T11, T23, and T38 isolates are genetically similar to French, Burundi, and Cameroonian strains, and T28 to French strains. The other HCV-4k Tunisian isolates do not have a strong closely related strain.
The cluster 1b includes five Tunisian strains. T18, T20, and T26 seem to have a relationship with European strains (Germany, Greece, and Spain), and T22 and T25 are closely related to Anglo-Saxon strains (UK, USA, Ireland, and Australia).
Discussion
Hemodialysis patients are one of the high risk groups for hepatitis, especially HCV infection (30,34,35). In Tunisian dialysis patients, the prevalence of infection varies from 43% before 1995 (17) to 19.07% in 2002 (3), and this infection was correlated with a history of blood transfusions and with a long-term hemodialysis. Hepatitis C viral infection varies widely among dialysis units all over the world (34). This variation is due to several factors such as blood transfusion, intravenous drug users (IVDU), and lapses of universal precautionary measures. In Tunisia, epidemiological and previous phylogenetic studies conducted on both healthy and HCV infected subjects, reported a large predominance of genotype 1b (4,10,12,26). Few epidemiological studies have focused on HCV-4 infection in Tunisia.
Several molecular methods have been used for genotype HCV; nucleotide sequencing of a phylogenetically informative region remains the gold standard (5,16).
In this study, the genotyping was done in the first time for the same samples by reverse line probe assay (LiPA; INNO-LiPA HCV II; Innogenetics) of the 5’UTR, and in the second time by sequencing of the DNA of the NS5b region. The results by the two techniques are concordant for 28/33 of cases (84.8%). However, for the other 5 isolates (15.2%) typed genotype 4 by INNO-LiPA, their direct sequencing in the NS5b region identified a genotype 1b. This discordant result is confirmed by the sequencing of these 5 isolates in the constant region 5′UTR. These results corroborate those of Scott et al. (41) who reported that, while most assays target the highly conserved 5’ noncoding region of the HCV genome, mutations can occur in this region leading to misclassification of HCV genotypes in 5%–8% of cases. Similarly, the study of Chen et al. (8) showed that the reverse line probe assay LiPA was not heterogeneous enough for use in determination of HCV subtype and cannot be used for differentiation of HCV genotypes 1a and 1b.
The HCV genotype 4 has 19 assigned subtypes (a-h and k-u) (24). This genotype is highly prevalent in the Middle East (Teheran, Yemen, Kuwait, Iraq, and Saudi Arabia) and in Africa, particularly in Egypt due to the use of unsterile equipment during mass treatment of the population with parenteral antischistosomal therapy from the 1920s to the 1980s(7,13,15,18). HCV-4 has recently spread in several Western countries, especially in Europe (28,33) and North America, due to the variations in population structure, immigration, and routes of transmission, particularly among IVDU populations, who represent the main reservoir for HCV in Europe. In this study, phylogenetic analysis revealed the presence of three different clusters of HCV4. The most prevalent subtype of HCV-4 circulating among the dialysis centers in Tunis region, were 4k. In fact, the subtype 4k submerged the Tunis city. The analysis of these strains according to their original dialysis unit seems to indicate that the F public dialysis center could be the source of contamination. So, all patients whose strains belonged to cluster 4k passed through the F public center for a mean hemodialysis duration ranging from a few months to several years before joining their respective private center. Even if most Tunisian HCV-4k isolates were unknown, three of them seem to be related with African strains (Burundi and Cameroon), although these strains are close to French strains. Since human exchanges between Tunisia and these two African countries are rare, the most likely hypothesis is that these strains were transmitted by people traveling between Africa and France.
A second cluster grouped only 15.2% of the strains identified as HCV-4a that are related to the sequences described in Egypt, where a high prevalence of HCV-4 has been described and approximately 63% of Egyptian isolates belong to HCV-4a (1,43). These results corroborate those reported by Djebbi et al. (11).
The third cluster contains one isolate that is strongly related to Egyptian strain HCV4o, and six others isolates weakly related to HCV4o strains. These isolates appear to represent a new subtype. They should be analyzed in another region of the viral genome, especially in the hyper variable region E1/E2 which, according to many authors, would best reflect the full extent of subtype variations present in the infected individuals (16) or better yet by sequencing the whole genome (24,44).
In conclusion, this study showed that blood transfusions are common major risk factors of infection, especially concerning the HCV contamination in hemodialysis units. The analysis of the phylogenetic tree revealed, as previously reported, that the HCV-1b strains were closely related to Anglo-Saxon and European isolates, while the HCV-4 isolates are genetically similar to Egyptian and African strains. The HCV4k is the most prevalent subtype of HCV-4 circulating among the dialysis centers in the Tunis region, and a new subtype of HCV4 seems to be suspect in this city.
Footnotes
Acknowledgments
This study was supported by a grant from the Tunisian Kidney Transplantation Research Fund (LR03SP01) and MEDIS Laboratory. We thank all the medical staff and technicians of dialysis centers who agreed to participate in this study. We are grateful to Professors Alain Goudeau and Catherine Gaudy, University François Rabelais, Microbiology Laboratory, Tours (France) for our initiation to the phylogenetic analysis methods for the study of the HCV diversity.
Author Disclosure Statement
No benefits in any form have been received or will be received from commercial party related directly or indirectly to the subject of this article.