
Abstract
Major histocompatibility complex (MHC) class II expression is critical for the presentation of antigens in the immune response to viral infection. Consequently, some viruses regulate the MHC class II-mediated presentation of viral antigens as a mechanism of immune escape. In this study, we found that Coxsackievirus B3 (CVB3) infection transiently increased IK expression, which reduced the expression of MHC class II (I-A/I-E) on splenic B cells. Interestingly, CVB3-induced IK elevated cAMP, a downstream molecule of the G protein-coupled receptors, which inhibited MHC class II presentation on B cells. Transgenic mice expressing truncated IK showed lower expression of MHC class II on B cells than did wild-type mice after CVB3 infection. Taken together, these results imply that IK plays a role in downregulating MHC class II expression on B cells during CVB3 infection through the induction of cAMP.
Introduction
Coxsackievirus B3 (CVB3) is one of the picornaviruses, with a single-stranded positive-sense RNA genome (24). CVB3 has been associated with myocarditis and dilated cardiomyopathy (17,24,52). A previous study reported that the myocytes of CVB3-infected mice induced MHC class I but not MHC class II (44). Moreover, although the mechanisms underlying the adaptive immune response to CVB3 have been previously reported (13,23,41), the role and mechanism of MHC class II in CVB3 infection are yet to be determined (17). To clarify the mechanism of the CVB3-mediated immune response, we performed a microarray assay with infected mouse hearts and demonstrated the expression of several interesting genes, including IK (unpublished data).
IK contains a trimeric coiled-coil motif, a nuclear localization signal (NLS), and a region of exclusive tandem repeats of Arg (R)–Asp (D) and Arg (R)–Glu (E), which is commonly found in RNA-binding proteins (2). It has been reported that IK is an efficient inhibitor of IFNγ-induced MHC class II expression and it was originally isolated from the conditioned culture medium of the K562 erythroleukemic cell line (25). IK has also been implicated in the inhibition of constitutive MHC class II expression through the repression of class II transactivator (CIITA) (33,49), a factor that regulates MHC class II expression (10,43). However, the detailed cellular mechanism of IK action remains to be clarified. One study showed that the truncated IK (tIK) protein, which includes the 242 amino acids from residue 316 (methionine) to residue 557 (tyrosine), is incapable of localizing in the nucleolus because the 315 amino acid residues are deleted from the N-terminus of the full-length IK protein, but still functions in reducing MHC class II expression (2,33). Therefore, it has been suggested that tIK affects MHC class II expression through its interaction with several unknown cellular factors in the cytoplasm (49). However, the cellular factors that act as adaptors are still unknown. Although the regulation of MHC class II plays an important role in antiviral immunity, how IK is involved in the MHC class II mediated antiviral response is still poorly understood.
In this study, we investigated the role and cellular mechanism of IK during CVB3 infection. We found that CVB3 infection transiently induces IK and downregulates MHC class II expression on B cells. A focus on the mechanism of IK revealed that it interacts with some G protein-coupled receptors (GPCRs) and induces intracellular cyclic AMP (cAMP), ultimately inhibiting MHC class II expression on B cells. Taken together, our results suggest that IK supports, at least partly, viral immune escape in splenic B cells during the early stage of CVB3 infection.
Materials and Methods
Wild-type and transgenic mice and cells
Male BALB/C strain mice (4 to 10 weeks old) purchased from Orient Bio (Seongnam, Korea). Wild-type mice and PCR-negative littermates were used as the controls. The transgenic founder mice were mated with wild-type BALB/C mice to produce the F1 generation. The F1 transgenic lines were mated with wild-type BALB/C mice to produce F2 mice, which were used for the phenotypic analyses. All animals were housed under specific-pathogen-free conditions and handled according to protocols for animal care approved by the Catholic University of Korea. Burkitt's lymphoma-derived human Raji B cells were maintained in RPMI 1640 medium with 10% fetal bovine serum (FBS; Hyclone Laboratories, Logan, UT), 2 mM
Generation of tIK transgenic mice
To generate truncated-IK-expressing transgenic (tIK-Tg) mice, the pronuclei of fertilized eggs from BALB/C mice were microinjected with the pcDNA– Hemagglutinin (HA)–tIK plasmid and implanted into BALB/C females. The construction of pcDNA–HA–tIK is described in the Materials and Methods. Female founders were mated with wild-type BALB/C males. tIK-Tg mice were identified by genotyping PCR for tIK performed on tail DNA at 3 weeks of age. The primer set for the genotyping PCR was: sense, 5′-CCCTATTGACGTCAATGACGGTAAATGGCC-3′; and antisense, 5′-CCGTCCTTCAGACATCTTGATGCCATACTG-3′.
Viral infection
The mice were infected by the intraperitoneal injection of 1×106 plaque-forming units (PFU) of CVB3 in 100 μL of virus stock diluted with serum-free medium (or an equal volume of serum-free medium as the control). Their organs were harvested 3–5 days after infection for RNA extraction, quantitative real-time reverse transcription–PCR (RT–PCR) analysis, and flow-cytometric analysis. Raji B cells were washed with serum-free medium and infected for 1 h with CVB3 at a multiplicity of infection (MOI) of 10. Following infection, the Raji B cells were placed into the wells of a six-well plate.
Isolation of splenic B cells
Mouse splenic B cells were obtained from BALB/C or tIK-Tg mice by magnetic cell sorting. To isolate the splenic B cells, spleen tissue was passed through a 70 μm cell strainer (BD Biosciences, San Jose, CA) and centrifuged. After several rinses with cold PBS, the splenocytes were washed in red blood cell (RBC) lysing buffer containing 8.3 g/L ammonium chloride in 0.01 M Tris-HCl buffer (pH 7.5±0.2) until no RBCs were visible. After RBC lysis, the splenic B cells were separated by negative selection with a magnetic non-B-cell-antibody cocktail using a MACS magnetic separation column (Miltenyi Biotec, Bergisch Gladbach Germany).
cDNA preparation from Raji B-cell total mRNA
Total mRNA from Raji B cells was isolated with TRIzol Reagent (Invitrogen, Carlsbad, CA). A total of 1×106 Raji B cells were harvested. After the cells were washed with PBS, the total mRNA was extracted and cDNA was synthesized using RT&GO (MP Biomedicals, Solon, OH) and an oligo (dT) primer (Cosmogenetech, Korea), according to the suppliers' instructions. The endogenous IK gene was identified by RT-PCR, using the primer sets: IK sense, 5′-CACCACGTGGCTGTTCTTAG-3′ and antisense, 5′-TCTTAGCATGTGGGACACAGA-3′; β-actin sense, 5′-CTCGTCGTCGACAACGGCTC-3′ and antisense, 5′-AAACATGATCTGGGTCATCTTCTC-3′. These primer sequences were autonomously obtained by Primer3 input program (version 0.4.0. on the WWW for general users and for biologist programmers).
Transient transfection with plasmid DNA containing the tIK gene or siRNA
The construction of plasmid DNA containing the tIK gene was as follows. We cloned cDNA of tIK from the tissues of CVB3-infected mice. Truncated IK (nucleotides 946–1674 of IK) was inserted into the pcDNA 3.1(+) vector (Invitrogen, Carlsbad, CA) with HA expression sequence. The plasmid was designated pcDNA–HA–tIK. To transfect the plasmid DNA containing the tIK gene, 20 μg of DNA was electroporated into Raji B cells (1×107 cells in 500 μL of RPMI 1640) with a Gene Pulser (BioRad, Hercules, CA) at 250 V, 950 μF, and ∞ Ω, in a 0.4 cm cuvette. After incubation on ice for 10 min, RPMI 1640 medium supplemented with 10% FBS was added to the cells, which were then incubated for 24 h. The cells were harvested 24 h after infection. Silencing IK gene expression with siRNA has been described previously (7). Purified splenic B cells (5×105 cells per mouse) were transfected with 150 pmol of the IK-targeting siRNA oligonucleotide (Dharmacon, Waltham, MA). The sequences were as follows: sense, 5′-TpsGpsTCTTCAAAAATATTCATGTCpsApsG-3′; and antisense, 5′-TpsCpsTCTTCTTTTCCTCTTCTCTCTCpsTpsC-3′ (7). At 24 h after transfection, the siRNA-transfected splenic B cells were infected with CVB3 for 48 h. The cells were harvested 48 h after infection.
Promoter assay
Raji B cells were cotransfected with 10 μg of reporter plasmid containing a luciferase gene under the control of the MHC class II promoter (MHCII–Luc), kindly supplied by Dr Chang (10). The effector plasmid (15 μg; pcDNA–HA–tIK or empty pcDNA 3.1(+) plasmid) and 5 μg of Renilla-luciferase-expressing plasmid (Promega, Madison, WI) are used (10). At 24 h after transfection, the cell lysates were assayed for firefly luciferase and then Renilla luciferase for normalization, using a dual luciferase assay kit (Promega), according to the manufacturer's instructions. Luciferase activity was calculated by dividing the firefly luciferase activity by the Renilla luciferase activity for each sample.
Yeast two-hybrid assay
A yeast two-hybrid assay was performed using the PBN204 yeast strain, as described previously (16). The cDNA library was from human T cells and was purchased from Clontech (Mountain View, CA). All Y2H-positive colonies were assayed with PCR amplification of the corresponding plasmid inserts using the T7 sense primer, 5′-TAATACGACTCACTATAGGGCG-3′ and the AD antisense primer, 5′-AGATGGTGCACGATGCACAG-3′. The PCR products were purified and used as the templates for DNA sequencing per Y2H-positive clone. The primer set for the sequencing reactions was: sense, GAL4-ADF, 5′-TACCACTACAATGGATGATG-3′; and antisense, pGAL4 AD-3, 5′-GAACTTGCGGGGTTTTTC-3′.
Plaque assay
The titers of the infectious viruses were determined by plaque assay of the individual homogenates of organs, prepared with an HT10 homogenizer (IKA-Korea, Korea). The homogenates were serially diluted 10-fold and then inoculated onto HeLa monolayers in 6-well plates. The plates were incubated for 1 h at 37°C in 5% CO2. After virus absorption, complete serum-free Dulbecco's modified Eagle's medium (DMEM) supplemented with 2% FBS and containing 0.5% agar was added to each well. The plates were incubated at 37°C in 5% CO2 for 2 days. After incubation for 1 day, the cells were fixed with methanol:ddH2O:acetic acid (45:45:10) for 5 min at room temperature and the agarose gel discarded. The cells were then stained with crystal violet solution (Sigma, St. Louis, MO) for 10 min at room temperature and washed with water. Plaques were counted with the naked eye and viral titers were calculated as plaque-forming units (PFU) per gram, as described previously (45).
Plaque reduction neutralization test
Sera of CVB3-infected WT and tIK-Tg mice were serially diluted two fold from 1:10 to 1:1280 with serum-free medium. The virus–antibody mixture was prepared by mixing 100 PFU of CVB3 with the serially diluted serum samples, and incubated at 37°C for 1 h. Aliquots (100 μL) of the virus–antibody mixture were inoculated onto HeLa monolayers in 6-well plates. The plates were incubated for 1 h at 37°C in 5% CO2. After virus absorption, complete serum-free medium (DMEM) supplemented with 2% FBS and containing 0.5% agar was added to each well. The plates were incubated at 37°C in 5% CO2 for 2 days. The cells were then fixed with methanol:ddH2O:acetic acid (45:45:10) for 5 min at room temperature and the agarose gel discarded. The cells were stained with crystal violet solution (Sigma, St. Louis, MO) for 10 min at room temperature and washed with water. Plaques were counted with the naked eye.
Quantitative real-time RT–PCR
Total RNA was extracted from the cells with TRIzol Reagent (Invitrogen, Carlsbad, CA), according to the manufacturer's instructions. Equal amounts of DNase-treated RNA were used for cDNA synthesis. cDNA was synthesized with RT-&GO (MP Biomedicals, Solon, OH) and RT–PCR was then performed. Real-time PCR was performed on a MyiQ-single iCycler (BioRad) using the SYBR Green PCR Master Mix (Takara Bio Inc., Japan). The expression of a given gene was quantified with the comparative threshold cycle (ΔCT) method and normalized to a mouse housekeeping gene (encoding 18S rRNA). The primer set for IK was: sense, 5’-CACCACGTGGCTGTTCTTAG-3’; and antisense, 5’-TCTTAGCATGTGGGACACAGA-3’. The primer set for CVB3 mRNA was: sense, 115-AGTAACACACTCCGATCAACA-135; and antisense, 208-GAGCAGTCTATTGATACTCAGTCC-185. All oligonucleotides were synthesized by CosmoGenetech (Korea).
Flow cytometry
Cells were infected with CVB3 and some CVB3-infected Raji B cells were treated with 100 ng/mL pertussis toxin (PT; Calbiochem, Darmstadt, Germany), a cAMP-elevating toxin, for 12 h. A total of 1×106 Raji B cells were harvested. After the cells were washed in ice-cold phosphate-buffered saline (PBS) containing 1 % bovine serum albumin (BSA), they were stained for cell surface-expressed HLA-DR with phycoerythrin (PE)-labeled monoclonal antibody (mAb) directed against human HLA-DR and an isotype control (all from BD Biosciences, San Jose, CA) on ice for 30 min. The cells were then washed three times with PBS containing 1% BSA and resuspended in 1% paraformaldehyde. To stain splenic B cells for MHC class II (I-A/I-E) and CD45R/B220, the cells were incubated with anti-I-A/I-E mAb (PE-labeled clone 2G9), anti-CD45R/B220 mAb (FITC-labeled clone RA3-6B2), or the appropriate isotype controls (all from BD Biosciences, San Jose, CA). The stained cells were analyzed using a FACScan flow cytometer and Beckman analysis software (Beckman Coulter, Inc., Brea, CA).
Intracellular cAMP measurement
Raji B cells were transferred into 12-well plates after transfection with tIK-encoding plasmid and cultured in RPMI medium supplemented with 10% FBS and 1% penicillin–streptomycin. On the next day, the cells were infected with virus as described above. cAMP was quantified using a cAMP assay kit (GE Healthcare, UK). The assays were conducted according to the supplier's instructions.
Statistical analysis
The experimental values are expressed as mean±SD. Significance between groups was evaluated by one-way ANOVA, followed by the Tukey multiple range test for post hoc comparison of group means or Student t test. Differences between groups were considered significant at p<0.05.
Results
CVB3 infection transiently increased IK expression and reduced MHC Class II expression in mouse splenic B cells
In a microarray analysis of CVB3-infected mouse hearts (unpublished data), we demonstrated the induction of IK expression by CVB3 infection. To focus on the B-cell immune response after CVB3 infection, we isolated splenic B cells and measured the levels of IK mRNA by quantitative real-time RT–PCR and observed that IK mRNA was upregulated in CVB3-infected mice 3 days post infection (pi) (Fig. 1A). These results are consistent with the microarray data that showed that IK was induced in CVB3-infected mouse hearts (Supplementary Fig. S1; supplementary material are available at
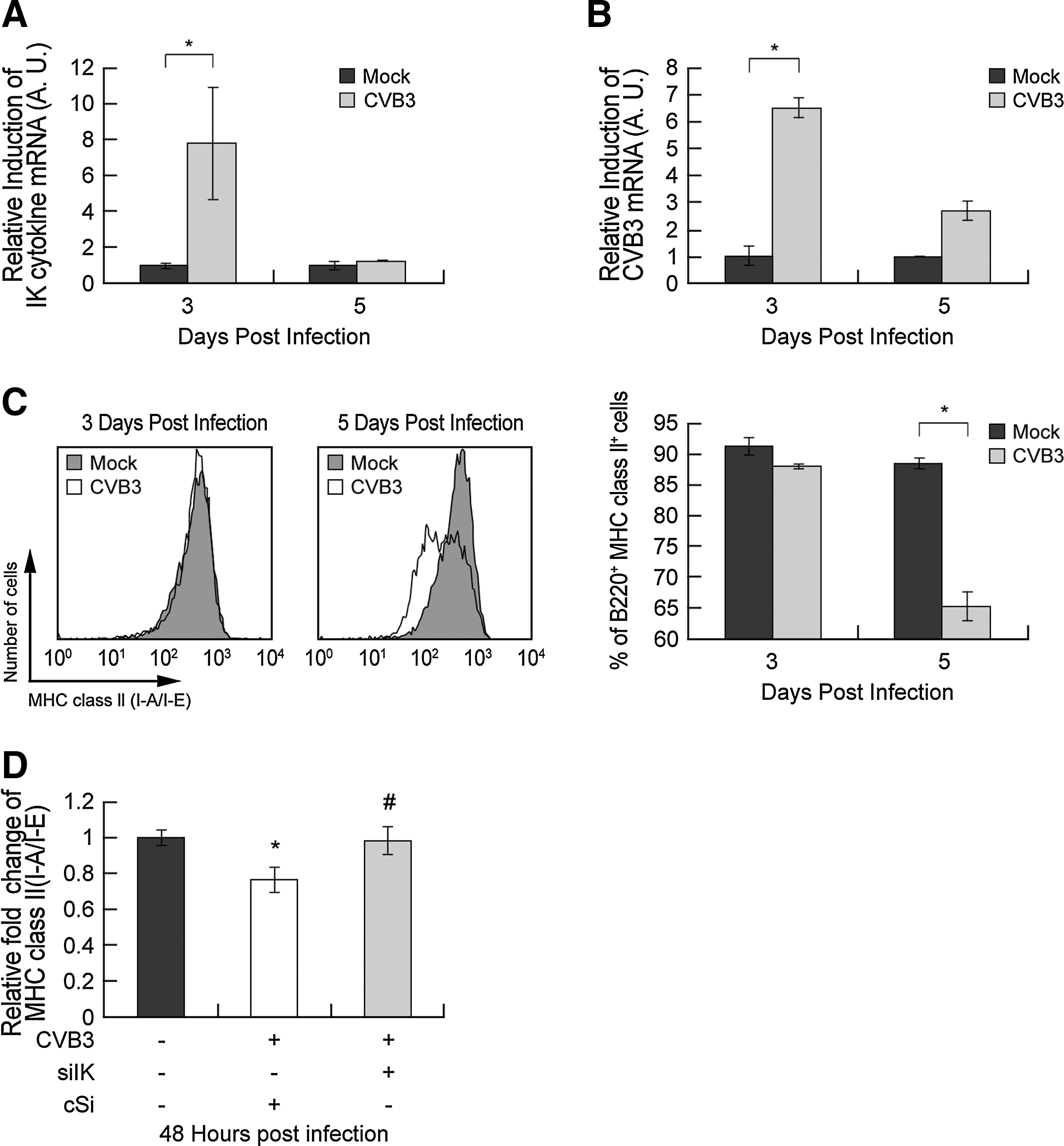
CVB3 infection transiently increased IK and reduced MHC class II expression in splenic B cells.
tIK reduced MHC class II expression on CVB3-infected Raji B cells
Raji B cells are a human Burkitt's lymphoma cell line (38). Interestingly, Raji B cells do not express endogenous mRNA encoding IK protein (49). We confirmed this by RT-PCR (Fig. 2A). Therefore, we used this cell line as the endogenous-IK-gene-negative cells to analyze the role of IK. A previous study showed that truncated IK (tIK), encoded by a 0.72-kb fragment of IK (residues 316–557), is functional in transfected cells (33). Furthermore, the treatment with exogenous tIK protein caused a reduction in IFNγ-induced MHC class II expression in murine macrophage cell lines (33). In this study, to analyze the role of tIK in MHC class II regulation during CVB3 infection, we constructed a plasmid encoding Hemagglutinin (HA)-tagged tIK, designated pcDNA–HA–tIK (Fig. 2B) and assessed the expression of tIK protein with anti-HA antibody (Fig. 2C). When pcDNA–HA–tIK was transfected into Raji B cells, which were then infected with CVB3, a flow cytometric analysis showed that tIK downregulated MHC class II expression during CVB3 infection (Fig. 2D). CVB3 infection usually upregulates MHC class II (HLA-DR) expression in human Raji B cells, which lack an endogenous IK gene (Fig. 2A). However, compared with Raji B cells transfected with the control vector, pcDNA–HA–tIK-transfected cells showed reduced endogenous MHC class II expression, and the induction of MHC class II by CVB3 infection was also inhibited in these cells 24 h after CVB3 infection (Fig. 2D). We also performed an MHC class II promoter assay to show that tIK regulates MHC class II expression at the transcriptional level. We cotransfected Raji B cells with pcDNA–HA–tIK together with a plasmid expressing a luciferase protein under the control of the MHC class II promoter (MHC II–Luc) and Renilla-luciferase-expressing plasmid (pRL–null) for expression compensation, and then infected them with CVB3. The promoter activity, represented by the expression of luciferase, increased in the CVB3-infected Raji B cells (second column in Fig. 2E, 6.4×106 RLU/mL) but decreased by 2-fold in the CVB3-infected pcDNA–HA–tIK-transfected Raji B cells (fourth column in Fig. 2E, 3.3×106 RLU/mL). The pcDNA–HA–tIK-transfected Raji B cells also showed 2-fold reduction in MHC class II promoter activity when treated with IFNγ (second and fourth columns in Fig. 2F, 7.2×106 RLU/mL and 3.7×106 RLU/mL, respectively), an inducer of MHC class II expression (38). These data suggest that transfected tIK can reduce both endogenous MHC class II and CVB3/IFNγ-induced MHC class II. Moreover, the class II transactivator (CIITA) has previously been shown to be a key regulator of IFNγ-induced MHC class II and the MHC class II pathway accessory molecules (10,14,32,38,42). Therefore, to investigate the regulation of CIITA by tIK, we measured the levels of CIITA mRNA by quantitative real-time RT–PCR and observed that CIITA mRNA was downregulated in tIK-transfected Raji B cells at 24–48 h after transfection (Supplementary Fig. S2A). These data are consistent with previous studies (33,49). We also identified a reduction in the mRNA levels of other MHC class II accessory molecules, Ii chain and HLA-DM, with the quantitative real-time RT–PCR analysis of tIK-transfected Raji B cells at 24–48 h after transfection (Supplementary Fig. S2B and S2C). These data indicate that tIK inhibits CIITA and other targets of CIITA that affect MHC class II expression. Furthermore, BJAB B cells, which also lack an endogenous IK gene, showed similar responses to those of the Raji B cells, in so far as CVB3 infection and IFNγ treatment increased MHC class II promoter activity, whereas pcDNA–HA–tIK transfection reduced MHC class II activity (data not shown). Taken together, our data suggest that the exogenous expression of tIK protein in B cells inhibits not only constitutive MHC class II expression but also the induction of MHC class II expression during CVB3 infection and after IFNγ treatment by regulating the activity of the MHC class II promoter.
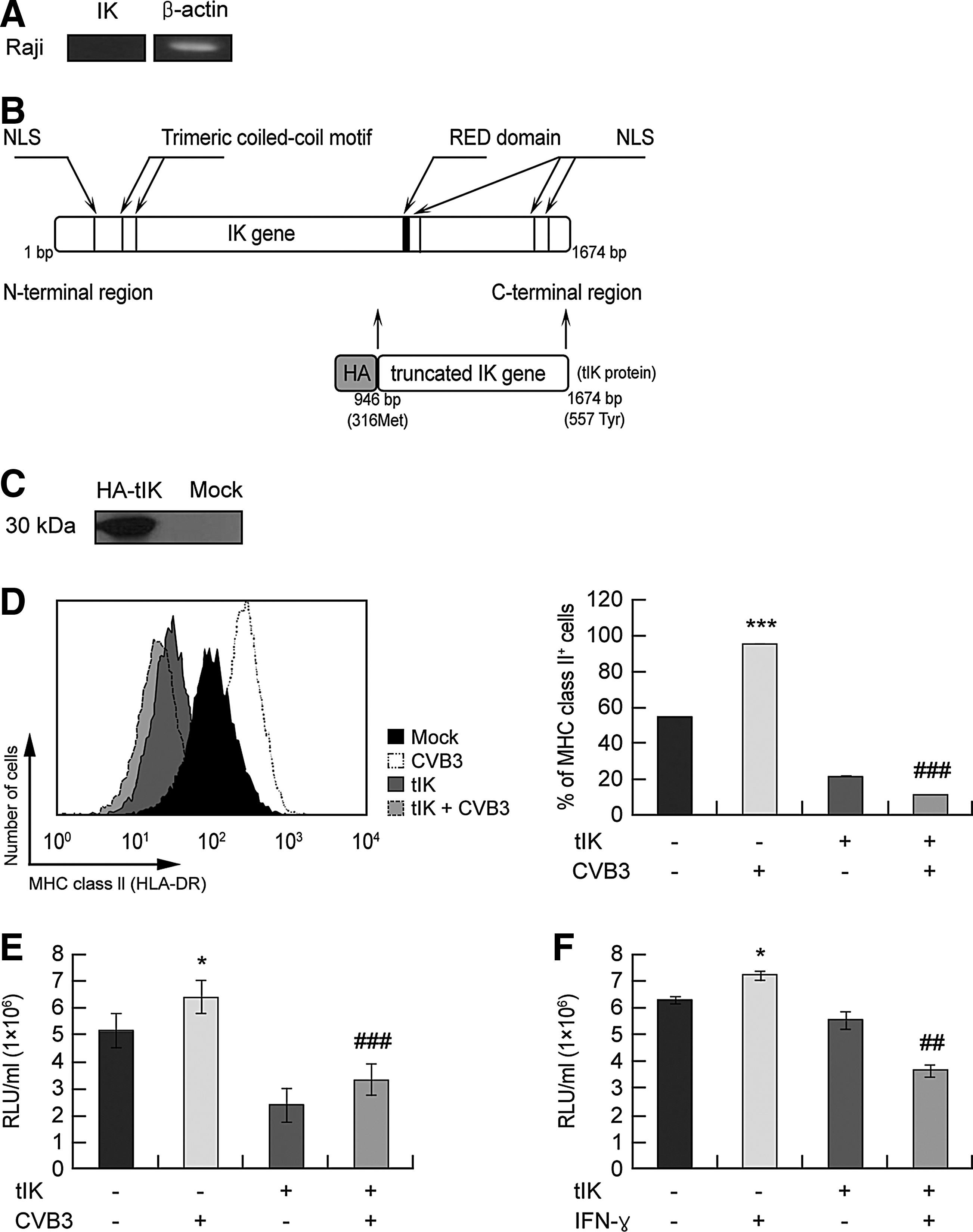
tIK reduced MHC class II expression in CVB3-infected human Raji B cells.
Identification of the tIK-binding protein families by yeast two-hybrid assay
CIITA is a key regulator of MHC class II expression (10,14,32,38,42). Previous studies have demonstrated that IK inhibits MHC class II expression through the indirect suppression of CIITA transcription (49). To identify the possible cellular mechanisms underlying the effects of tIK protein, we conducted a yeast two-hybrid assay using tIK protein as the bait. This test revealed several cellular tIK-binding factors, such as protein kinase, carrier protein, receptor protein, adhesion molecule, and G protein-related protein (Fig. 3). Furthermore, G protein-coupled receptors (GPCRs) constituted a high proportion of the G protein-related protein group. Collectively, these data suggest that tIK protein binds to some GPCRs and is related to the G protein-mediated signaling pathway.
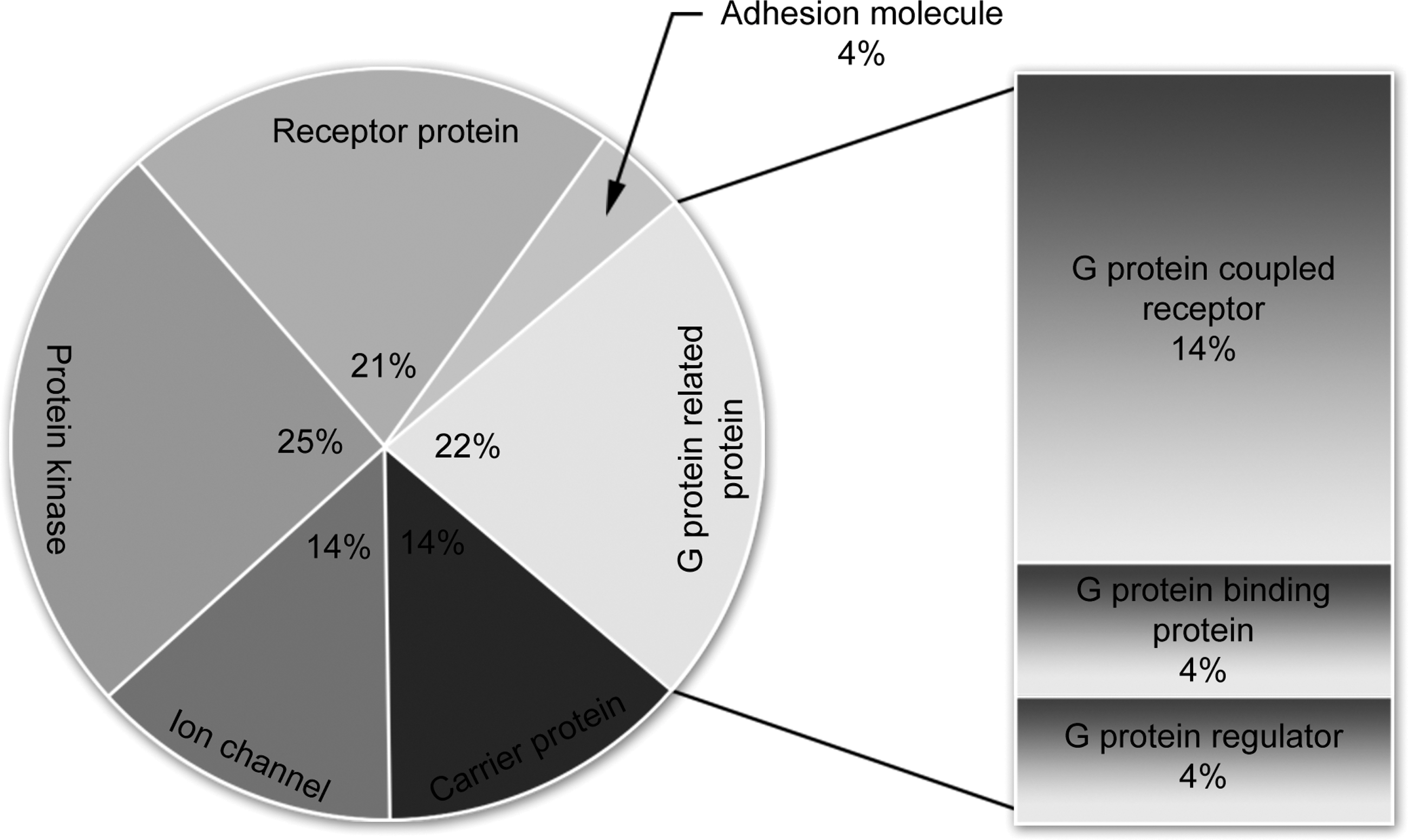
Identification of tIK-binding protein family with a yeast two-hybrid assay. A yeast two-hybrid assay (Y2H) was used to determine the tIK–cellular factor interactions. Y2H was performed using the PBN204 yeast strain with URA3, LacZ, and ADE3 reporters and the GAL promoter. PCR amplification was performed on all Y2H-positive colonies and the PCR products were purified and used as the templates for sequencing the genes encoding the tIK-binding proteins.
tIK reduced MHC class II in CVB3-infected Raji B cells by elevating intracellular cAMP
To further define the effects of the interaction between GPCR and tIK protein, we quantified the intracellular cAMP levels, because cAMP is a general downstream molecule of GPCRs (5). The cAMP levels were three-fold higher in the pcDNA–HA–tIK-transfected Raji B cells than in the mock-vector-transfected Raji B cells after 24 h (Fig. 4A). In contrast, CVB3 infection reduced cAMP levels compared with the control level (noninfected cells). However, pcDNA–HA–tIK transfection increased cAMP levels, even in CVB3-infected cells (Fig. 4B). To examine the effects of cAMP on MHC class II expression, we next treated CVB3-infected Raji B cells with PT (purtussis toxin). PT is known to play an important role in the modulation of the immune system as a multiple suppressor and cause elevated cAMP levels (8,12), which ultimately reduces MHC class II (HLA-DR) expression on the surfaces of monocytes, thus suppressing the serum antibody response (9,46). We also verified that treatment with PT alone elevated cAMP levels in Raji B cells (Supplementary Fig. S3A). CVB3-infected Raji B cells showed increased MHC class II (HLA-DR) expression. However, PT-treated CVB3-infected Raji B cells showed lower MHC class II expression than that on non-PT-treated CVB3-infected control cells at 24 hours pi (Fig. 4C). We also checked the viability of the Raji B cells after PT treatment using an WST-1 assay kit (Roche), and found that PT treatment did not affect cell viability (Supplementary Fig. S3B). To confirm the role of cAMP in CVB3-regulated MHC class II expression, as well as increasing cAMP with PT, we directly treated CVB3-infected Raji B cells with dibutyryl–cAMP, a cell-permeable analogue of cAMP that activates cAMP-dependent protein kinase (11,31). As when CVB3-infected Raji B cells were treated with PT, dibutyryl–cAMP-treated CVB3-infected Raji B cells showed reductions in MHC class II expression and CIITA mRNA at 24 h pi relative to those in untreated CVB3-infected cells, although the effect was not as dramatic as that produced with PT (Supplementary Fig. S4A and S4B). Thus, cAMP negatively regulates MHC class II expression on B cells. Collectively, our results indicate that CVB3-induced IK increases cAMP levels via GPCRs, and increased cAMP downregulates MHC class II expression.
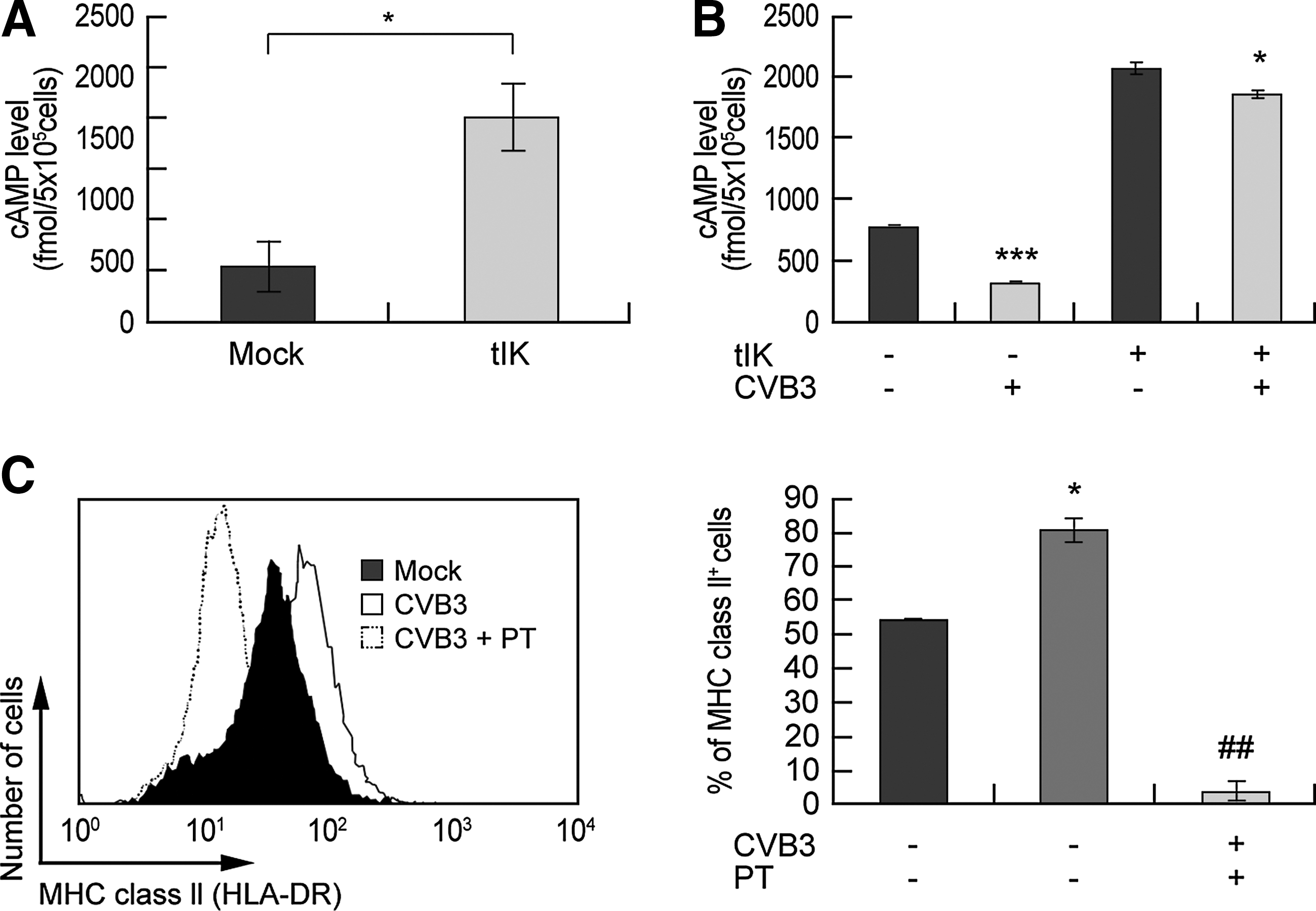
tIK reduced MHC class II expression in CVB3-infected Raji B cells by elevating intracellular cAMP.
Transgenic mice expressing tIK showed reduced MHC Class II expression on splenic B cells during CVB3 infection
To confirm the role of tIK protein in vivo, we generated mice transgenic for HA-tagged tIK in the BALB/C genetic background (tIK-Tg mice), and screened them by PCR using genotyping primers (Fig. 5A). Most of the screened mice were tIK heterozygous (tIK-Tg+/–), because very few tIK homogenic mice (tIK-Tg+/+) were generated (data not shown). Therefore, we used tIK-Tg+/– mice for further study. Although we used the cytomegalovirus promoter to express tIK in the transgenic mice, we found no difference in the steady-state expression of tIK mRNA between the tIK-Tg+/–mice and their PCR-negative littermates (data not shown). These findings are similar to those in other cytokine-transgenic mice (4,36). The behavior, growth, and reproduction of the tIK-Tg+/– mice were normal compared with those of their littermate controls. Moreover, there were no differences between the PCR-negative littermates and the wild-type mice (data not shown), and so both these groups were regarded as wild-type (WT) mice.
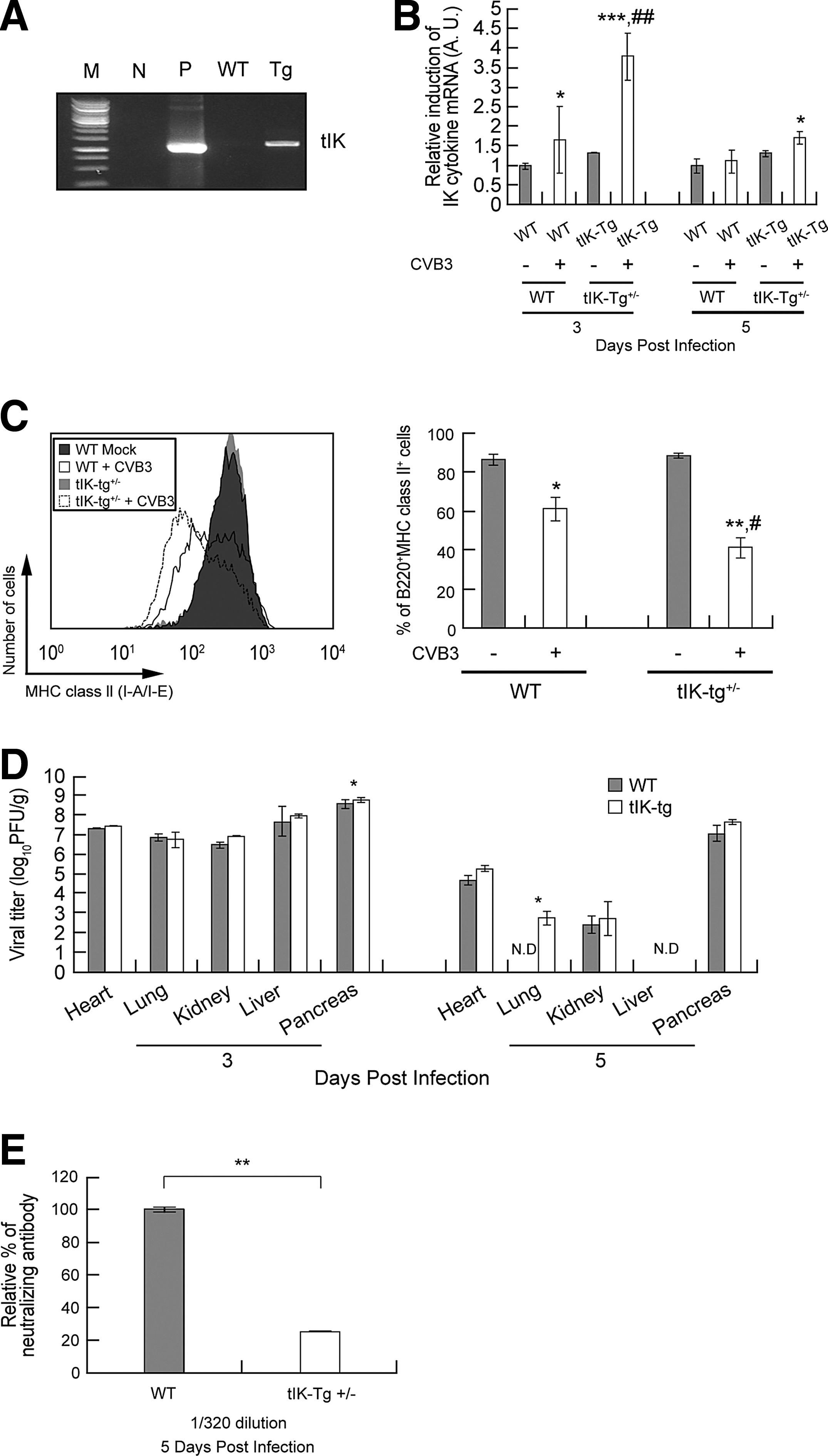
Transgenic mice expressing tIK showed reduced MHC class II expression on splenic B cells during CVB3 infection.
To determine the effects of CVB3 infection on tIK protein in the splenic B cells of tIK-Tg+/– mice, we infected tIK-Tg+/– mice and WT mice with CVB3. The splenic B cells isolated from the tIK-Tg+/– mice showed higher IK mRNA levels than those from WT mice 3 days after CVB3 infection (Fig. 5B). Interestingly, and as expected, the splenic B cells of the CVB3-infected WT mice showed reduced MHC class II expression, whereas the splenic B cells of the CVB3-infected tIK-Tg+/– mice showed a synergistic effect, with a further reduction in MHC class II expression compared with that in the cells of the CVB3-infected WT mice (Fig. 5C). We also examined the function of IK in the downregulation of MHC class II expression and consequently in the generation of neutralizing antibodies, because MHC class II presentation is closely related to antibody production (9,39). The tIK-Tg+/– mice showed similar viral titers to those of the WT mice in various organs at 3 and 5 days pi, except that the viral titer in the lung at 5 days pi in the tIK-Tg+/– mice was a little higher than that in the WT mice (Fig. 5D). The CVB3-infected tIK-Tg+/– mice also showed an approximately four-fold reduction in the relative percentage of neutralizing antibodies compared with that in the CVB3-infected WT mice (Fig. 5E). Thus, the reduction in neutralizing antibodies in the tIK-Tg+/– mice did not result from a reduction in the viral titer. We considered the possibility that a reduction in CD4+ T cells affected the production of neutralizing antibodies (26), but found that the numbers of CD4+ T cells after CVB3 infection did not differ in the WT and tIK-Tg+/– mice. Although the number of CD4+ T cells tended to be higher in the tIK-Tg mice than in the WT after CVB3 infection, this difference was not statistically significant (Supplementary Fig. S5). Based on the data discussed above (Fig. 5D and 5E, and Supplementary Fig. S5), the reduction in neutralizing antibodies may be attributable to the reduction in MHC class II expression induced by tIK.
Taken together, these data suggest that CVB3-induced IK did not affect viral growth (except in the lung at 5 days pi), but downregulated MHC class II expression, ultimately affecting the production of neutralizing antibodies.
Discussion
This study demonstrates that IK is a factor that regulates MHC class II expression in B cells during an early stage of CVB3 infection. CVB3 is reported to induce myocarditis, which later develops into dilated cardiomyopathy (17,24). A previous mouse study showed that MHC class I (H-2Kk) antigens are strongly induced by CVB3 in cardiac myocytes, whereas low or undetectable levels of MHC class II (I-Ak) antigens were observed (44). However, the mechanism of this response to CVB3 remains unknown. To investigate it, we conducted a microarray analysis and demonstrated the induction of IK mRNA in CVB3-infected mice. IK has been identified as a key regulator of constitutive and inducible MHC class II expression in a B cell line, through its interaction with several unknown cellular factors (25,49). Therefore, we hypothesized that this induced IK transiently downregulates MHC class II expression during CVB3 infection to delay the host immune response, allowing viral growth. Viruses can regulate the presentation of viral antigens on MHC class II molecules by reducing the expression of MHC class II in host cells, because MHC class II expression is essential for the presentation of antigen in the adaptive immune response (15). Viruses escape the host immune response in several ways, including by the inhibition of IFNγ–JAK–STAT signal transduction, which induces MHC class II expression, and the expression of a viral protein that affects MHC class II degradation and mis-sorting (15). Consequently, viruses can replicate in host cells temporarily undetected by CD4+ T cells (15,51). However, the role and mechanism of MHC class II downregulation via CVB3-induced IK had not been established.
In this study, we investigated how CVB3 infection affects MHC class II expression and how IK downregulates MHC class II expression. First, we observed in mice that CVB3 infection upregulates IK expression in splenic B cells and downregulates MHC class II expression on the surfaces of splenic B cells (Fig. 1). Second, we demonstrated the role of IK in MHC class II regulation during CVB3 infection in vitro. The exogenous expression of tIK protein (a truncated functional form of IK protein) hindered the induction of MHC class II expression by CVB3 infection or IFNγ treatment in endogenous IK-negative Raji B cells (Fig. 2D and 2E). Moreover, in tIK-Tg+/– mice, MHC class II expression was further downregulated on splenic B cells during CVB3 infection (Fig. 5C). Third, we identified an IK-mediated cellular mechanism that downregulates MHC class II expression on B cells during CVB3 infection. According to a yeast two-hybrid assay (Fig. 3), IK is associated with the GPCR family expressed on the B-cell surface, which is previously published the presence of GPCR family on B-cells surface (19,34,40). However, it must be determined whether tIK engages directly with GPCRs or interacts with them indirectly. Therefore, more research is required to define the interaction between tIK and GPCRs. cAMP has previously been reported to be one of the main regulators of MHC class II expression (20,21,27), and elevated cAMP levels cause the downregulation of MHC class II expression via the regulation of CIITA expression (3,6,53). Moreover, the downregulation of MHC class II expression and the reduction of neutralizing antibodies have been attributed to cAMP-elevating reagents, such as PT (8,9,12,46). We observed that tIK transfection increased the levels of cAMP, a molecule downstream from GPCRs (5), in B cells, which ultimately reduced MHC class II expression (Fig. 4). This suggests that IK regulates MHC class II expression through the upregulation of cAMP levels. Eventually, tIK-Tg+/– mice showed low levels of neutralizing antibodies compared with those in WT mice, induced by CVB3 infection (Fig. 5). Therefore, the reduction of MHC class II expression by IK affects the production of neutralizing antibodies during CVB3 infection, even when the viral titers are similar.
A previous article showed that CVB3 infection completely and selectively inhibited antigen presentation via the MHC class I pathway, whereas presentation of CVB3 epitopes via the MHC class II pathway is inhibited to a lesser extent (22). There are two discrepancies between this study and another article and our data. The first is related to the inhibition of MHC class I. Seko (44) reported that CVB3 infection strongly induced MHC class I but not MHC class II in cardiac myocytes. Our unpublished data show that CVB3 infection strongly triggered T cell infiltration into myocytes. Considering that these infiltrated T cells may be mostly activated CD8+ T cells, T cells may be activated in some way after CVB3 infection. More detailed experiments are required to confirm T cell activation by CVB3 infection. The second issue is the inhibition of MHC class II. Our data do not show complete and permanent inhibition of the MHC class II pathway; rather, our data show only that CVB3 infection temporally inhibited the induction of MHC class II via IK expression in the early infection stage and that, as a consequence, CVB3 infection can trigger the MHC class II pathway after the disappearance of IK expression. In other words, these CVB3-induced IK data simply showed the events in the early stage that help the virus evade the host immune response. Therefore, similar to the findings of a previous study (22), our data suggest that CVB3-induced IK is inhibited transiently in the early infection stage but that the induction of MHC class II is eventually restored.
Taken together, our data demonstrate the regulation of the adaptive immune response to CVB3 infection. It has previously been reported that CVB3 infection induces IFNγ, which affects antibody production by enhancing MHC class II expression, as well as virally induced inflammation (18,28,30,35,37). We also found that CVB3 upregulates IFNγ in the splenic B cells of WT mice (Fig. 6A). Therefore, we deduced that CVB3 downregulates IFNγ-induced MHC class II expression by inducing IK expression at an early stage of infection. In this way, CVB3 can regulate the adaptive immune response of its host by balancing CVB3-induced IFNγ and IK expression, allowing its replication in the host (Fig. 6A and 6B). Although these findings increase our understanding of IK-regulated MHC class II regulation by CVB3 in B cells, we must also confirm the relationship between IK and MHC class II in other antigen-presenting cells, such as macrophages and dendritic cells. Moreover, if we can identify the mechanism of IK in this immunoregulation, it may be possible to find a therapeutic use for IK in the reduction of MHC class II expression in autoimmune diseases.
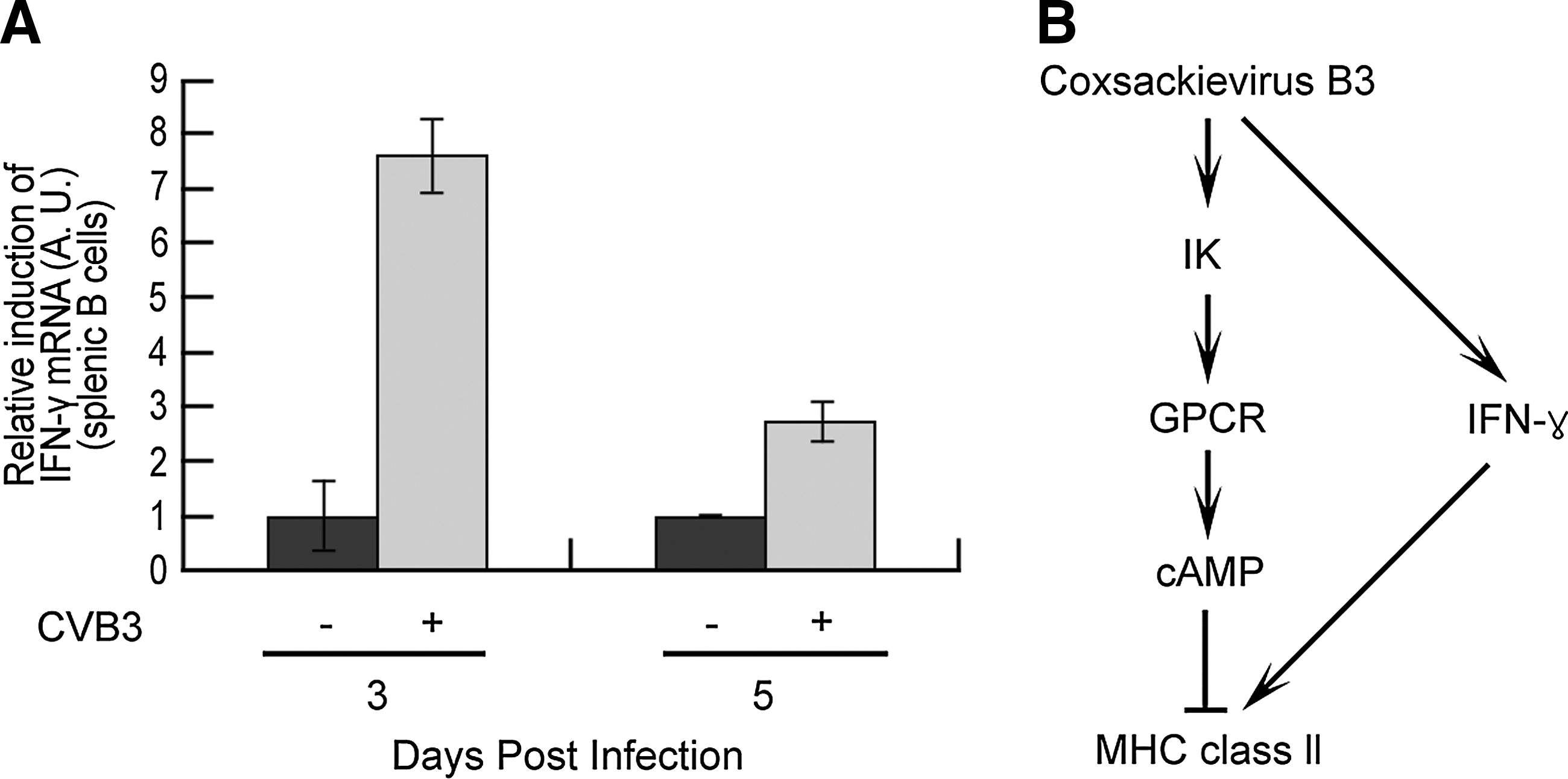
Scheme for the regulation of MHC class II expression by balancing CVB3-induced IFNγ and IK.
Footnotes
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (no. 2009-0076644) and by the Catholic University of Korea, Research fund, 2011. Hye-Lim Park was supported by a grant from the Health Fellowship Foundation of the Yuhan Corporation.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.