
Abstract
Swine influenza virus (SIV) is a fast-evolving viral pathogen in pig populations. However, commercial vaccines, based on inactivated viruses, cannot provide complete protection with induced humoral immunity only and require frequent updates to fight against current isolates. A DNA vaccine delivering conservative epitopes was designed in this study in the hope of meeting the need. In this study, a B-cell epitope (HA2.30-130), a quadruplicated Th-cell epitope (NP55-69), and a quadruplicated CTL epitope (NP147-158) were fused separately to the C-terminal of VP22c gene in the modified pcDNA3.1 plasmid. The expression of epitopes was confirmed by in vitro transfection of 293FT cells. The DNA vaccine administered intramuscularly stimulated epitope-specific immunity against the two T-cell epitopes in all ten mice before the virus challenge. Only two out of ten mice were ELISA positive against the B-cell epitope. All vaccinated mice survived a lethal dose of virus challenge, while all mice in the challenge control group died. The DNA vaccine delivering epitopes in this study showed promising protection against influenza virus in an animal model; however, more work needs to be done to evaluate the best conserved protective epitopes which can be applied in developing a universal DNA vaccine.
Introduction
S
Current swine influenza vaccines in North America are inactivated whole-virus vaccines. They can induce only humoral immune responses, which fail to protect against mismatched strains (23). Broad-spectrum humoral and cellular immunity are essential for providing cross-protection against heterologous viruses, especially for fast-evolving influenza viruses (6,34). DNA vaccines can induce both humoral and cellular immunity (27). To achieve broad-spectrum immunity, epitope vaccines have been explored in order to develop a universal influenza vaccine (31); however, epitopes are too short to be immunogenic without an appropriate carrier or adjuvant. Inserting these epitopes into DNA vectors may be a potential way to reach the goal with the DNA-epitope vaccine platform established by our group previously (42).
Antibodies against the stalk region of HA molecules are neutralizing antibodies that inhibit the membrane fusion of virion and endosome instead of obstructing the binding of virus to the host cell (9,13,33,40). Several broad cross-reactive monoclonal antibodies target conformational epitopes in the stalk region at different positions and confer protection as both prophylactic and therapeutic treatments (10,25,33,40,45,46). This domain consists of three regions: the primary region is the N-terminal segment of HA2 (amino acid 38–55, 76–130), and the N- and C- terminal of HA1. Vaccines based on the headless HA or synthetic peptide HA2.76–130 protected mice against homologous and heterologous viruses (2,32,41). The Th-cell epitope NP55-6-9 can enhance and prolong immunity (16,18). The CTL epitope NP147–158 has been proven to confer protection against homo- and heterologous virus infection (7,8,16,18,35).
To develop an effective DNA-epitope vaccine against SIVs, multiple approaches were applied to optimize the immunogenicity of DNA vaccines: 1) enhancing antigen expression by applying codon optimization (39) and insertion of the Kozak sequence (17,26) and a chimeric intron (4,28); 2) increasing the number of target protein presenting cells by fusing the epitopes with the C-terminal portion of the VP22 gene (VP22c) from bovine herpesvirus-1 to enhance intercellular migration of fused proteins (50); 3) augmenting antigen presentation by quadruplicating the T-cell epitopes (19,48,49) spaced with linker -RVKR- targeting peptide presentation by MHC class I (20,21,24). This vaccine design was evaluated as a platform for developing DNA-epitope vaccines.
Materials and Methods
Mice
BALB/c mice at the age of 6–8 weeks were purchased from Harlan Sprague Dawley (Indianapolis, IN). The experimental protocol was approved by the Purdue Animal Care and Use Committee (PACUC).
Construction of DNA plasmids
Epitopes selected in this study were a B-cell epitope HA2.30–130 (QGSGYAADLKSTQNAIDGITNKV NSVIEKMNTQFTAVGKEFSHLERRIENLNKKVDDGFLDIWTYNAELLVLLENERTLDYHDSNVKNLYEKVRSQLKNNA), a Th-cell epitope NP55-69 (RLIQNSITIERMVLS), and a CTL epitope NP147-158 (TYQRTRALVRTG). The construction of pVP22cEpitope was referenced from Wei et al. (42). The two T-cell epitopes were quadruplicated and linked with linker -RVKR-; the genes were codon-optimized to species Mus musculus and synthesized by GenScript (Piscataway, NJ). The epitopes were fused to the C-terminal of VP22c (denoted as pVP22cHA2, pVP22cNP55, and pVP22cNP147).
In vitro transfection of DNA plasmids
The 293FT cells were cultured in complete DMEM as described previously (42). DNA plasmids at 1 μg were mixed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) at an N/P of 5 and transfected into 293FT cells. The plates were examined after 48 h incubation.
Immunofluorescence assay (IFA)
Anti-His (C-term) monoclonal antibody (Invitrogen) at a dilution of 1:200 was applied to acetone-fixed cells followed by fluorescein labeled goat anti-mouse IgG (H+L) (KPL, Gaithersburg, MD) at a dilution of 1:60. The staining was observed with Nikon Eclipse TE2000-S (Nikon instruments, Melville, NY).
Immunization procedures
There were three groups with 10 mice per group. Mice in group 1 (G1-NC) were sham inoculated with PBS buffer. Group 2 (G2-CC) were sham inoculated with PBS and challenged with virus as the vaccinated group. Group 3 (G3-IM) were vaccinated intramuscularly in the tibialis anterior muscles with 100 μg of the three-plasmid mixture (33.3 μg per plasmid) in 100 μL of volume. Mice were vaccinated three times at 3-week intervals and challenged with 10 LD50 of mouse-adapted SIV (A/swine/Indiana/67/2007 (H1N1)) at 2 weeks after the third dose. Half of the mice from each group were euthanized with CO2 before the virus challenge to measure the cellular immunity by flow cytometry. All the remaining mice were monitored closely for clinical signs and body weight until 14 days post virus infection (DPI). The mice were euthanized and considered dead when they were moribund and had more than 20% body weight drop (NIH recommended procedure). All the surviving mice by 14 DPI were euthanized. Serum was collected via submandibular veins before the second and third dose of vaccine and before the virus challenge. Lungs were collected from each mouse after euthanization.
Intracellular cytokine staining assay (ICCS) for IFN-γ
This assay was performed as described previously (42). Peptide NP147–158 was synthesized by Genscript (Genscript, Piscataway, NJ). NP55–69 and HA2.30–130 were synthesized by Almac (Almac Group Ltd, Craigavon, Northern Ireland). IFN-γ-secreting CD4+ or CD8+ T cells were detected as an indicator of cellular immunity.
Epitope-specific ELISA and microneutralization assay
Epitope-specific ELISA and microneutralization (MN) assay were conducted as previously described (42). The epitope-specific antibody levels were reported as the mean OD450 of each group. Samples were considered positive when the mean ODs of the samples were twice as high as the mean of the negative controls. MN titer was defined as the highest dilution that completely inhibited virus infection.
Virus isolation (VI) and quantitative real-time PCR (qPCR)
Lungs were homogenized in phosphate buffered salt solution (PBSS) at a final concentration of 10% (w/v) using a tissue homogenizer. Viral RNA was extracted with MagAttract Virus Mini M48 Kit (Qiagen, Valencia, CA) with a KingFisher instrument (Thermo Fisher Scientific, Inc., Waltham, MA). Real-time PCR for SIV H1N1 (29) was used to quantify the amount of viral genomic copies in the lung sample. SIV isolation from lung homogenate was conducted according to “WHO Manual on Animal Influenza Diagnosis and Surveillance” (44).
Statistical analysis
One-way ANOVA followed by pair-wise comparison with Tukey's test was applied to the data analysis. A p value of<0.05 was considered significant.
Results
Confirmation of the DNA plasmids constructs
DNA constructs were shown in Figure 1A schematically. The expression of pVP22cEpitope was successfully visualized in transfected cells by detecting 6XHis tag as shown in Figure 1B.
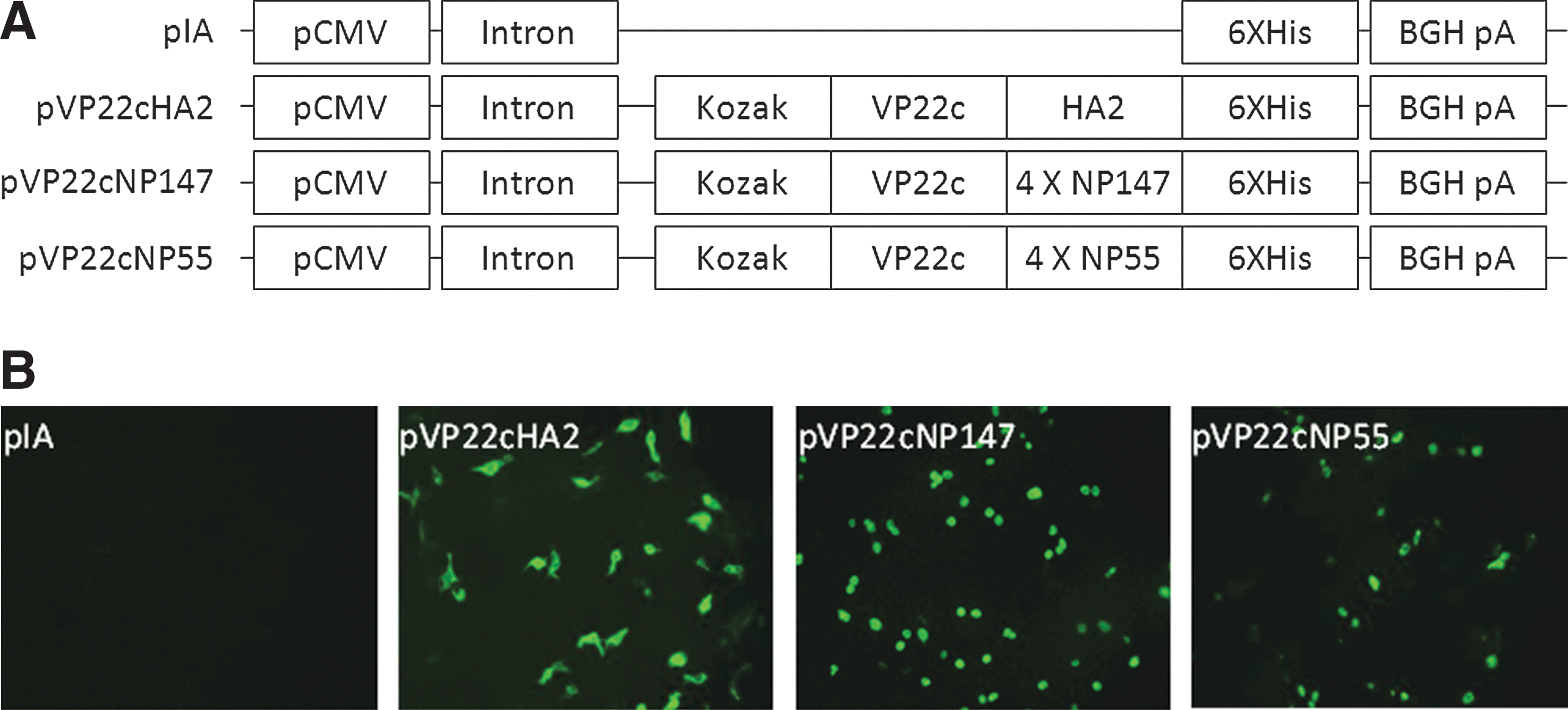
Construction of plasmids and transfection.
Intracellular cytokine staining assay (ICCS) for IFN- γ
The cellular immune response upon vaccination was indicated by detecting the IFN-γ-expressing cells with flow cytometry. The group mean percentage of epitope-specific IFN-γ+ lymphocytes is shown in Figure 2. In this study, NP147–158-specific IFN-γ+CD8+ lymphocytes and NP55–69-specific IFN-γ+CD4+ lymphocytes were detected in all the mice in G3-IM, which were statistically different from the control groups. The INF-γ-secreting cells were not detected in any groups upon stimulation of splenocytes with peptide HA2.30–130 (data not shown).
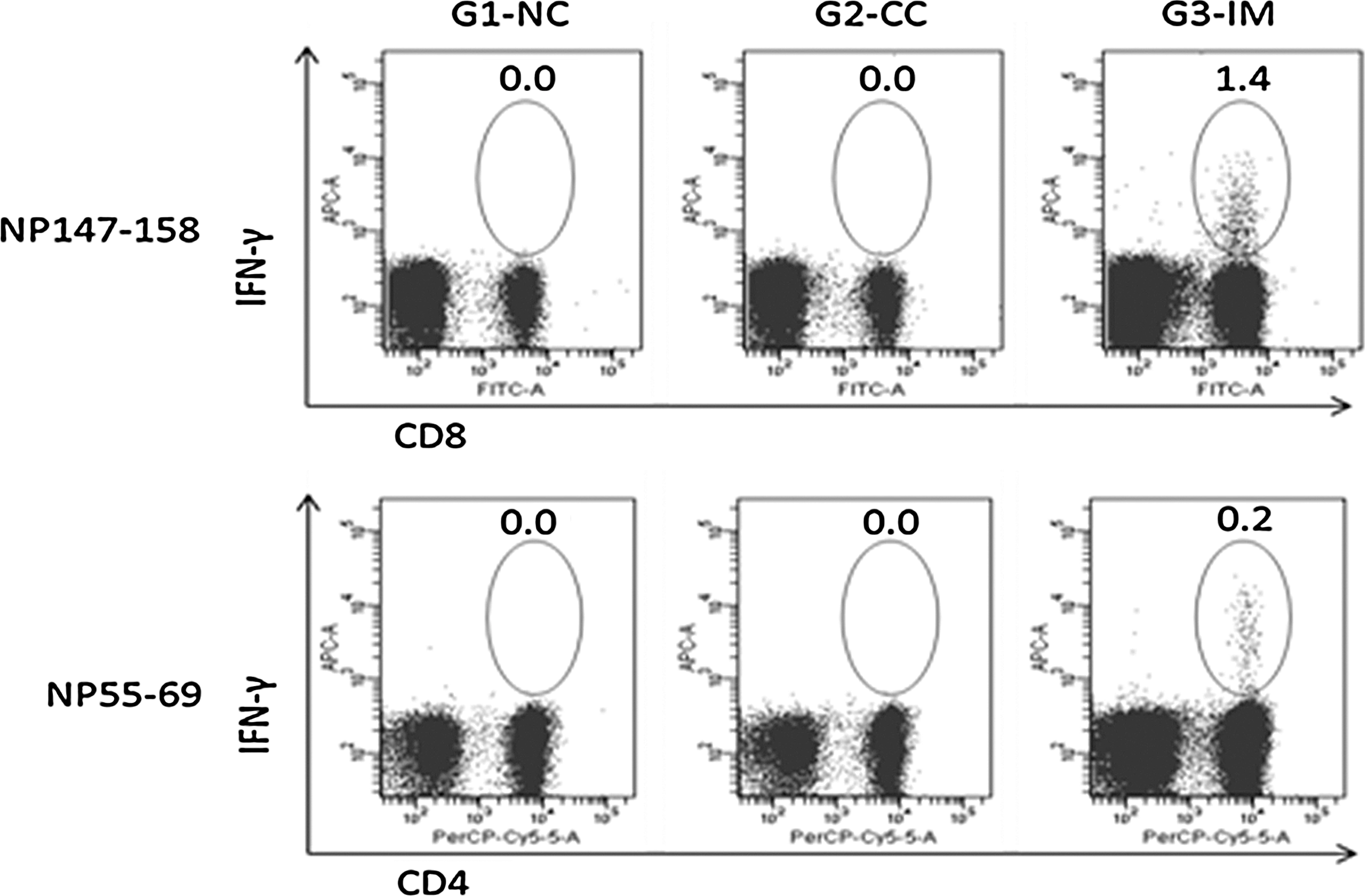
Flow cytometric analysis of the percentage of epitope-specific for IFN-γ secreting lymphocytes. Numbers in figures show the group mean percentage upon in vitro stimulation of splenocytes with synthesized epitopes. NP147–158-specific IFN-γ+CD8+ cells and NP55–69-specific IFN-γ+CD4+ cells were detected in G3-IM only, and the group mean as shown in figures is statistically higher than controls (p<0.001).
Humoral immune response
Epitope-specific humoral immunity was detected by ELISA (data not shown). In G3-IM, 1/10 was positive against peptide HA2.30–130 after the second dose and 2/10 were positive against this peptide after the third dose and before the virus challenge. No mouse in the other groups was positive for this peptide. No antibody response was detected against peptides NP55–69 and NP147–158 in any group. The serum samples collected before the virus challenge were tested for neutralizing antibodies by MN. No neutralizing antibody titer was detected in any serum sample (data not shown).
Mice survival curve and body weight drop
The mice survival curve and the body weight change after virus infection is shown in Figure 3. The mice in G1-NC remained healthy through the study. All the mice in G2-CC started showing respiratory disease at 2 DPI. They were severely affected with dramatically dropped body weight and no mice survived by 7 DPI. All the mice in G3-IM experienced mild body weight drop and survived until the end of the study.
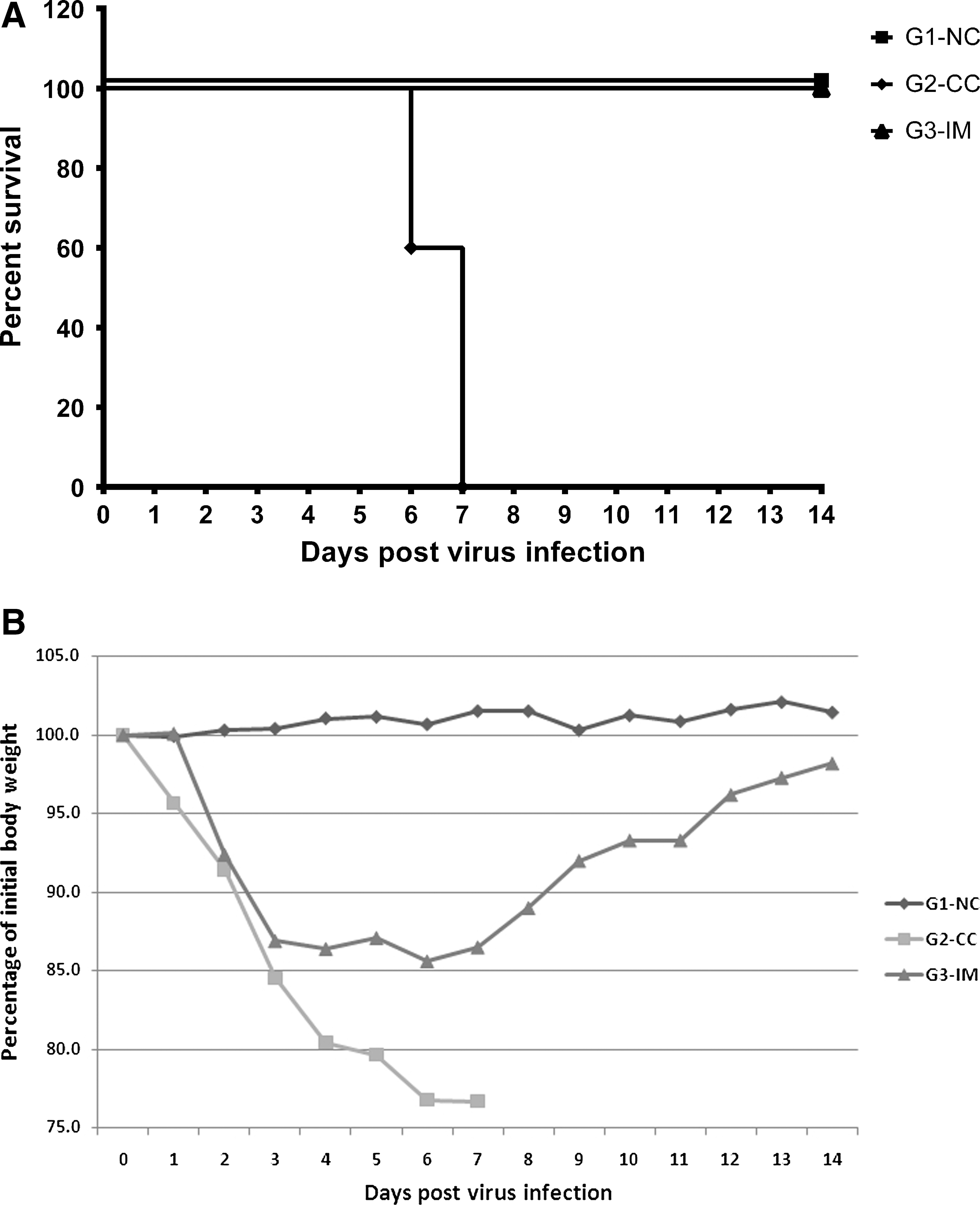
Protective efficacy of developed vaccine.
Virus isolation and quantitative real-time PCR
Virus isolation (VI) and quantitative real-time PCR (qPCR) were conducted on lung homogenate. No SIV was isolated from the mice in G3-IM and negative control group G1-NC euthanized at 14 DPI and no virus was detected by qPCR (data not shown). The mice in G2-CC, who developed severe clinical signs and had to be euthanized during earlier post virus infection according to NIH guidelines, were positive for VI and had very high viral load in lung samples ranging from 104 to 105 viral genome copies per mg of lung tissue.
Discussion
Current influenza vaccines are not capable of conferring protection against emerging variants. Herein, we explored the feasibility of designing a universal influenza DNA vaccine that encodes multiple conserved B- and T- epitopes among at least H1 subtype of swine influenza viruses.
T-cell immunity has been shown to mediate protection against influenza virus infections via direct transfer of CTL to infected mice (15,22,36,43,47). Studies in immunoglobulin knockout mice showed that mice were still able to clear virus infection but less effectively than normal mice (1,3,11,12). In this study, the mice in G3-IM were protected from death that is most likely correlated with the positive cellular immunity elicited by the two T-cell epitopes. The role of humoral immunity is questionable in this group. No antibody response could be detected against the two T-cell epitopes in this study, which further confirms that the two epitopes are T-cell epitopes. Studies with either depletion of T or B cells or immunization with a single epitope should be able to explain which one provided the protection to mice.
T-cell epitopes located in internal proteins are more conserved and serve as targets to stimulate broad-spectrum heterosubtypic immunity. Prime and boost infections with influenza viruses with different subtypes showed that T-cell immunity is the one that provided protection against heterologous virus infections (1,5,14,15). Therefore, T-cell epitopes are essential in vaccine design aiming to protect animals against broad influenza virus infections. In this study, only a homologous virus challenge was conducted. Studies using heterologous viruses to conduct the challenge should be able to prove if the two T-cell epitopes can serve as universal vaccine candidates.
In this study, no HA2.30–130-specific cellular immune response was detected, which might suggest that there is no T-cell epitope within this region. HA2.30–130-specific antibody response was detected only in 2/10 mice in G3-IM even after three doses of vaccination and before the virus challenge. In our previous study with pVP22cM2e (containing four copies of M2e per plasmid) as one of the plasmids in G2, there was an M2e-specific antibody response detected after three doses of plasmids at 25 μg per dose (42). It might be feasible to increase the immunogenicity by inserting multiple copies of HA2.30–130 in the plasmid backbone to increase the expression level in vivo. The serum samples collected from mice before the virus challenge were titrated for neutralizing antibody; however, no neutralizing antibody was detected even in the two ELISA-positive serum samples. This might be due to the low sensitivity of this assay or the HA2.30–130-specific antibodies could not neutralize the virus due to incorrect antigen presentation after in vivo expression. Headless HA molecule (2,32) and synthetic peptide HA2.76–130 (41) have been tested as vaccines in experiments. Two vaccine studies using the headless HA molecule mimicked the conformation of HA stem region. A vaccine study using synthetic HA2.76–130, eliminating other regions in the HA stalk domain, achieved broader protection compared to the former vaccines. The expressed peptide by HA2.30–130 in this study might form a differed three-dimensional conformation compared to the region of HA2.76–130, which might lead to the hiding of protective epitopes in this region. Several broad cross-reactive monoclonal antibodies targeting the HA stem region were mapped to different amino acids in the same domain or different domains in HA molecules (9,10,25,33,40, 45,46).The region in the HA stalk domain should be mapped in great detail for its role in stimulating protective immunity.
In summary, the DNA vaccine design in this study can be utilized as an effective platform for screening protective epitopes. Cellular immunity can protect mice from death, but not from developing disease. Humoral immunity is required to protect animals against disease development. The combination of both B- and T- cell epitopes in this DNA vaccine design is a highly promising method to develop a universal vaccine that provides broad protection against both homo- and heterologous viruses.
Footnotes
Acknowledgments
We thank the Purdue University “Lew Runnels Funds” for funding this study. We also thank Yi-Ning Chen for assistance in this study.
Authors Disclosure Statement
No competing financial interests exist.