
Abstract
Estrogen has been shown to increase resistance to HIV/SIV transmission by increasing the thickness of the genital epithelium. The immunological role of estrogen in HIV infection of primary target cells is less well characterized. We have found that primary macrophages are a target for anti-HIV activity of 17β-estradiol (E2). E2 did not affect surface expression of CD4 and HIV co-receptors nor HIV attachment to monocyte-derived macrophages (MDMs). In addition, E2 treatment blocked infection by a co-receptor-independent HIV-1VSV-G pseudotyped virus. Quantitative polymerase chain reaction analysis of HIV reverse transcribed DNA products indicated that E2 blocked HIV reverse transcription. E2 upregulated gene expression of interferons (IFNs) in MDMs from multiple donors. However, induction of host restriction factors APOBEC3G, APOBEC3F, or SAMHD1 was not consistent, with exception of APOBEC3A. Anti-HIV activity of E2 was abolished in the presence of IFN-α neutralizing antibody, and was absent in bone marrow–derived macrophages from IFN-α receptor deficient mice. Interestingly, HIV overcame E2-mediated HIV inhibition by suppressing induction of IFNs when MDMs were exposed to HIV before E2 treatment. These results offer a new mechanism of E2 on HIV inhibition. Future studies on the interplay between HIV and E2-mediated innate immune responses will likely provide insights relevant for development of effective strategies for HIV prevention.
Introduction
G
Studies in macaques and humans have suggested that estrogen protects the host against HIV infection through enhancement of physical barriers at the genital mucosa by increasing the thickness of the genital epithelium. Estrogen induces thickening of the vaginal epithelium in postmenopausal women on hormone replacement therapy (14,33) and in ovariectomized macaques (44,45). Additionally, in humans, it also rapidly keratinizes the inner foreskin, the site for HIV entry in the penis (38), and may protect men against HIV. Postmenopausal women who have lower estrogen and thinner vaginal epithelium (36) are more susceptible to HIV transmission (European Study Group on Heterosexual Transmission of HIV, 1992).
In contrast to its effect on the genital epithelium, the immunological role of estrogen in HIV protection is understudied. Estrogen exerts both inflammatory and anti-inflammatory effects depending on various factors such as target cells, immune stimuli, microenvironment, and estrogen concentration (reviewed in (46)). Enhanced HIV replication has been found in ex vivo ectocervical tissue from postmenopausal women compared to tissue from premenopausal women, and this HIV enhancement is associated with increased inflammation (42). However, the effect of estrogen on HIV infection of specific primary target cells and its underlying mechanism has not been well defined.
Macrophages are one of the major HIV target cells present in the female genital mucosa (43). While estrogen generally suppresses production of pro-inflammatory cytokines including IL-6, IL-1β, and TNF-α by monocytes and macrophages (15,24,32), it promotes IFN-γ production (23) and TLR4-mediated pro-inflammatory cytokine production through activation of macrophages (4). In this study, we found that 17β-estradiol (E2) protected primary macrophage against HIV infection, and we demonstrate a novel mechanism of E2-mediated HIV inhibition through IFN-α induction.
Materials and Methods
Reagents
Recombinant human IL-2, mouse anti-human CD3 Ab (clone UCHT1), and mouse anti-human CD28 Ab (clone 37407.111) were purchased from R&D Systems (Minneapolis, MN). Mouse anti-human IFN-α mAb (clone MMHA-2) was obtained from PBL Interferon Source (Piscataway, NJ). Water-soluble E2, water-soluble progesterone (P4), Histopaque®-1077, Triton X-100, RPMI-1640 medium, fetal bovine serum (FBS), modified RPMI-1640 medium (phenol red free), charcoal-treated FBS, human AB serum, and phytohemagglutinin (PHA) were purchased from Sigma-Aldrich (St. Louis, MO). PerCP-conjugated mouse anti-human CD4 (clone RPA-T4) was purchased from Biolegend (San Diego, CA). APC-conjugated mouse anti-human CD184 (CXCR4, clone 12G5), APC-conjugated mouse anti-human CD195 (CCR5, clone 3A9), and mouse IgG1 and IgG2a isotype controls were purchased from BD Biosciences (San Jose, CA).
Cell isolation and culture
All primary cells were cultured with RPMI-1640 medium supplemented with 10% heat-inactivated FBS. Peripheral blood mononuclear cells (PBMCs) from normal healthy blood donors were isolated by Histopaque®-1077 gradient centrifugation. Monocytes were isolated from PBMCs by adherence in flasks coated with human AB serum. Adherent cells were maintained in Roswell Park Memorial Institute medium (RPMI) with 20% FBS for 7 days and allowed to differentiate into monocyte derived macrophages (MDMs). FACS analysis showed that more than 95% of cells were CD68+, a marker for differentiated macrophages. CD4+ T-cells were isolated from PBMCs or peripheral blood lymphocytes (PBLs) by negative selection using a CD4+ T-cell isolation kit II (Miltenyi, Auburn, CA). PBMCs or CD4+ T-cells were then activated by incubation with PHA (5 μg/mL) and IL-2 (50 IU/mL) or by immobilized anti-CD3 Ab (1 μg/mL)/anti-CD28 Ab (0.6 μg/mL) for 3 days. Activated cells were washed with PBS and incubated with IL-2 before use. Before hormone treatment, all primary cells were incubated for 24 h in phenol red-free RPMI-1640 containing 10% heat-inactivated, charcoal-treated FBS.
HIV-1 infection
Replication-defective HIV-1JR-FL or HIV-1vsv Env-pseudotyped luciferase expressing reporter viruses used in single-cycle infection assays were produced in HEK293T-cells by co-transfection of a plasmid encoding the envelope deficient HIV NL4-3 virus and luciferase reporter gene (pNL-Luc-R+E-; gift of N. Landau, New York University, New York, NY) along with a plasmid encoding HIV-1JR-FL or VSV G envelope as described previously (6,8). Cells (PBMC or CD4+ T-cells at 1×106 per sample or MDMs at 1×105 per sample) were infected with pseudotyped luciferase reporter viruses (8–10 ng HIV p24 per sample) for 2 h at 37°C. After washing off unbound virus, MDMs or PBMCs/CD4+ T-cells were cultured for 2 or 3 days, respectively, before lysis in passive lysis buffer (Promega, Madison, WI). Luciferase activity (in relative light units) was measured on a Glomax 20/20 luminometer (Promega).
For multiple-round infection assays, cells were infected with CCR5-using HIV-1BaL virus (Advanced Biotechnologies, Inc., Columbia, MD) or primary isolates (the UNAIDS Network for HIV isolation and Characterization, Division of AIDS, National Institute of Allergy and Infectious Diseases) at a multiplicity of infection (MOI) of 0.05 for 2 h. After washing off unbound virus, cells were cultured, and HIV-1 p24 levels in cell culture supernatant were measured at different time points after viral infection by enzyme-linked immunosorbant assay (ELISA; HIV-1 p24 ELISA kit; SAIC-Frederick, Inc., Frederick, MD).
MTS assay
MDMs were plated in 96-well plates at 10,000 cells per well. The cells were treated with various concentrations of E2 for 24 h. Substrate from the CellTiter 96® Aqueous One Solution Cell Proliferation Assay (Promega) was added per the manufacturer's instruction. The optical density was measured at 490 nm.
FACS analysis
MDMs were incubated at 4°C in ice-cold PBS for 30 min and then detached using cell scrapers. Surface expression of CD4, CXCR4, and CCR5 were determined by staining the cells with fluorochrome-conjugated specific Abs. Appropriate isotype controls were included in all assays. Stained samples were analyzed on a BD Accuri™ C6 flow cytometer. Results were analyzed with FlowJo (Tree Star, Inc., Ashland, OR).
Real-time reverse transcription polymerase chain reaction
Total RNA was isolated from cells using TRIzol® (Life Technologies, Carlsbad, CA). To synthesize first-strand cDNA, 1000 ng of total RNA, oligo d(T)16 (25 μg/mL) and dNTP (0.5 mM) were incubated at 65°C for 5 min and quick-chilled on ice. RT was performed at 42°C for 50 min using SuperScriptTM II (Life Technologies). The PCR contained cDNA equivalent to 30 ng of RNA input, 200 nM primer sets, and SYBR Green Master Mix (Qiagen, Valencia, CA), and was run in a StepOnePlus real-time PCR system (Invitrogen/Life Technologies). Each reaction was performed in triplicate. The primer sequences were: GAPDH forward (5′-AGG TGA CAC TAT AGA ATA CTC TCT GCT CCT CCT GTT CG-3′), GAPDH reverse (5′-GTA CGA CTC ACT ATA GGG AAC GAC CAA ATC CGT TGA CTC-3′); IFN-α forward (5′-CAC ACA GGC TTC CAG GCA TTC-3′), IFN-α reverse (5′-TCT TCA GCA CAA AGG ACT CAT CTG-3′); IFN-β forward (5′-GAG CTA CAA CTT GCT TGG ATT CC-3′), IFN-β reverse (5′-CAA GCC TCC CAT TCA ATT GC-3′); IFN-γ forward (5′-AGC TCT GCA TCG TTT TGG GTT-3′), IFN-γ reverse (5′-GTT CCA TTA TCC GCT ACA TCT GAA-3′); APOBEC3A forward (5′-TGG CAT TGG AAG GCA TAA GAC-3′), APOBEC3A reverse (5′-TTA GCC TGG TTG TGT AGA AAG C-3′); APOBEC3F forward (5′-TAC GCA AAG CCT ATG GTC GG-3′), APOBEC3F reverse (5′-GCT CCA AGA TGT GTA CCA GG-3′); APOBEC3G forward (5′-GGC TCC ACA TAA ACA CGG TTT C-3′), APOBEC3G reverse (5′-AAG GGA ATC ACG TCC AGG AA-3′); SAMHD1 forward (5′-CCA AGC GTC CCC GTT GCG AT-3′), SAMHD1 reverse (5′-TCA AAG CCA CCG CGC CTG AG-3′). PCR conditions included a 95°C denaturation for 10 min followed by 40 cycles of 95°C for 15 s and 56°C for 60 s. Quantification of PCR products was normalized according to the amount of cDNA by amplifying GAPDH. Relative quantification of gene expression was calculated by using a ΔΔCt (Ct, threshold cycle of real-time PCR) method based on signal intensity of the PCR according to the following formula: ΔCT=Ct18S rRNA−Cttarget, ΔΔCt=ΔCtcontrol−ΔCtE2, Ratio=2−ΔΔCt.
Quantitative real-time PCR analysis of HIV-1 DNA
Total DNA was extracted from HIV-infected MDMs using the QIAamp DNA Blood Mini Kit (Qiagen). The level of HIV RT products was determined by quantitative real-time PCR analysis. Each PCR reaction contained 100 ng total DNA, primers (200 nM each), and SYBR Green Master Mix (Qiagen). The primer sequences for HIV-1 early RT products were M667 (5′-GGC TAA CTA GGG AAC CCA CTG-3′) and AA55 (5′-CTG CTA GAG ATT TTC CAC ACT GAC-3′); the primers for HIV-1 late RT products were M667 and M661 (5′-CCT GCC TCG AGA GAG CTC CAC ACT GAC-3′) (53). Standard curve for early or late RT products was generated with 10-fold serial dilutions of pNL4-3.Luc.R-E- ranging from 101 to 108 copies. PCR cycling conditions included 95°C for 10 min, followed by 40 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s. Reactions were carried out and analyzed using Stratagene MX3005P real-time PCR system (Agilent, Santa Clara, CA). The detection limit of early or late RT DNA products was 10 copies.
Preparation of mouse bone marrow derived macrophages (mBMDMs)
Male C57BL/6 mice were sacrificed and the femur and tibia were collected by carefully trimming off muscles and tendons. The bones were rinsed in ice-cold 95% ethanol for 1 min and then in PBS. Both ends of the bones were cut off using sterile forceps and scissors. Bone marrow was gently flushed out with RPMI-1640 medium containing 10% FBS using a 5 mL syringe and 27½ gauge needle.
The bone marrow cells were plated at 1×107 cells/100 mm petri dish (bacteriology grade) in 10 mL RPMI-1640 medium with 10% FBS and 40 ng/mL murine M-CSF (Peprotech, Rocky Hill, NJ). The cells were incubated at 37°C for 4 days, and then the culture supernatant was removed and replaced with fresh RPMI-1640 medium containing 10% FBS and 20 ng/mL murine M-CSF. After differentiation for 5 days, bone marrow derived macrophages (mBMDMs) at 1×105 per well in a 48-well plate were cultured in hormone-free medium for 24 h. Cells were pretreated with or without E2 followed by HIV-1vsv infection.
Statistical analysis
Differences between data sets were analyzed by a two-tailed Student t-test and Wilcoxon's signed-rank tests (SAS v9.2; SAS, Inc., Cary, NC). A p value of <0.05 was considered significant.
Results
E2 inhibits HIV-1 infection of primary MDMs
To determine the effect of sex hormones on HIV infection of primary HIV target cells, activated PBMCs, CD4+ T-cells, or MDMs were pretreated with E2 or P4 at different concentrations for 24 h before exposure to CCR5 (R5)-using HIV-1JR-FL pseudotyped luciferase reporter virus. E2 or P4 was added back after 2 h viral exposure, and was present during the infection. E2 exhibited a moderate inhibitory effect on HIV infection of PHA-activated PBMCs and a slight enhancing effect on infection of PHA-activated CD4+ T-cells (Fig. 1 A and B). E2 had no effect on HIV infection of T-cell receptor (TCR)-activated CD4+ T-cells (Fig. 1C), of HeLa-CD4-CCR5 cells, or of THP-1 cell line-derived macrophages (data not shown for HeLa and THP-1 cells). In contrast to the results in PBMCs or CD4+ T-cells, E2 at physiological concentrations (19,41,48), 10 and 100 nM, significantly inhibited HIV infection of MDMs (88% inhibition; Fig. 1D). E2 also blocked HIV replication in MDMs by R5 and dual-tropic X4R5 primary isolates (Fig. 1E). In contrast to E2, P4 at 1–100 nM did not exert any effect on HIV infection of activated PBMCs, CD4+ T-cells, or MDMs.

17β-estradiol (E2) inhibits HIV-1 infection of primary monocyte-derived macrophages (MDMs) but not activated peripheral blood mononuclear cells (PBMCs) or CD4+ T-cells.
We further characterized anti-HIV activity of E2 in primary macrophages and found that pretreatment of MDMs with E2 was sufficient to block HIV infection (Fig. 2A). However, prolonged treatment of MDMs with E2 (24 or 48 h) but not short-term exposure (2 or 8 h) was required to block HIV infection (Fig. 2B). Additionally, the anti-HIV activity of E2 was not due to cytotoxicity (Fig. 2C). MDMs were highly sensitive to LPS, a TLR4 agonist, which inhibits HIV infection. To confirm that anti-HIV activity of E2 was not due to LPS contamination, we examined the role of TLR4 activation in E2-mediated HIV inhibition. TLR4 inhibitor (CLI-095) abolished HIV inhibition by LPS but not E2, indicating that the HIV inhibitory effect of E2 was unlikely due to LPS contamination (Fig. 2D).

Prolonged treatment of MDMs with E2 is required to achieve HIV inhibition.
HIV exposure abolishes E2-mediated HIV inhibition
We then determined whether E2 exhibited any effect on viral infection in HIV-infected MDMs. Our results indicated that E2 did not have a significant inhibitory effect on HIV infection of MDMs among eight different donors when MDMs were exposed to HIV prior to E2 treatment (Fig. 3A and B). Similar results were obtained when replication competent virus HIV-1BaL was used (Fig. 3C).
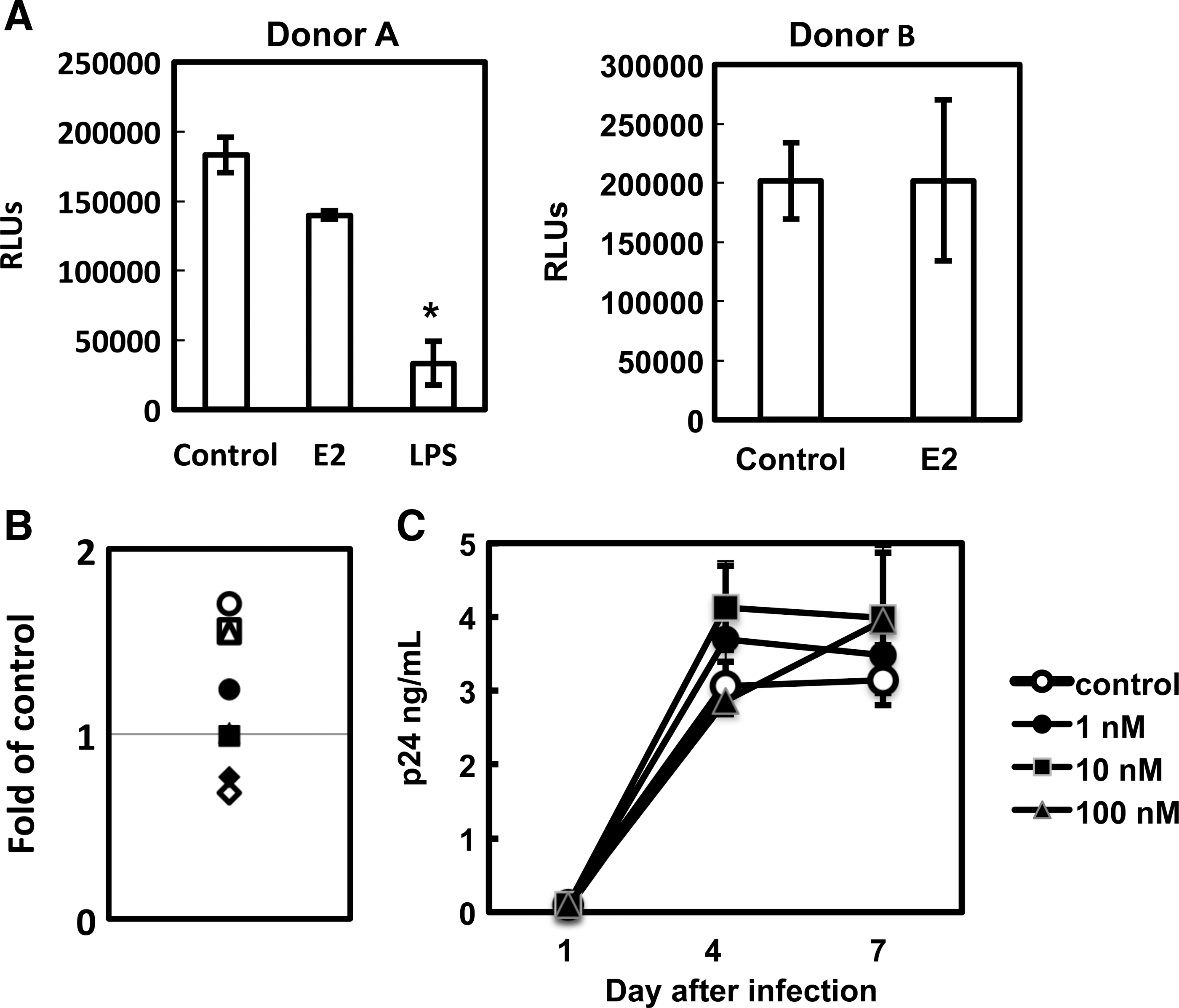
E2 does not inhibit HIV infection in HIV-infected MDMs.
E2-mediated HIV inhibition occurs after viral entry
To delineate the specific stages of the HIV life cycle that were blocked by E2, we first assessed whether pretreatment of MDMs with E2 affected HIV attachment. MDMs were treated with different concentrations of E2 for 24 h followed by exposure to HIV for 2 h at 4°C. After washing, cell-associated HIV p24 was determined by HIV p24 ELISA. There was no difference in HIV attachment in MDMs with or without E2 pretreatment (Fig. 4A). We also found that pretreatment of MDMs with E2 blocked HIV infection by pseudotyped virus with VSV G envelope, which is independent of CD4 and HIV co-receptors for viral entry (Fig. 4B). This result indicated that E2-mediated HIV inhibition was independent of HIV receptors. Additionally, FACS analysis of CD4 and HIV co-receptors demonstrated that E2 did not significantly alter cell surface expression of CD4, CXCR4, or CCR5 (Fig. 4C). Taken together, these results indicate that E2 blocks HIV infection at the postentry level.
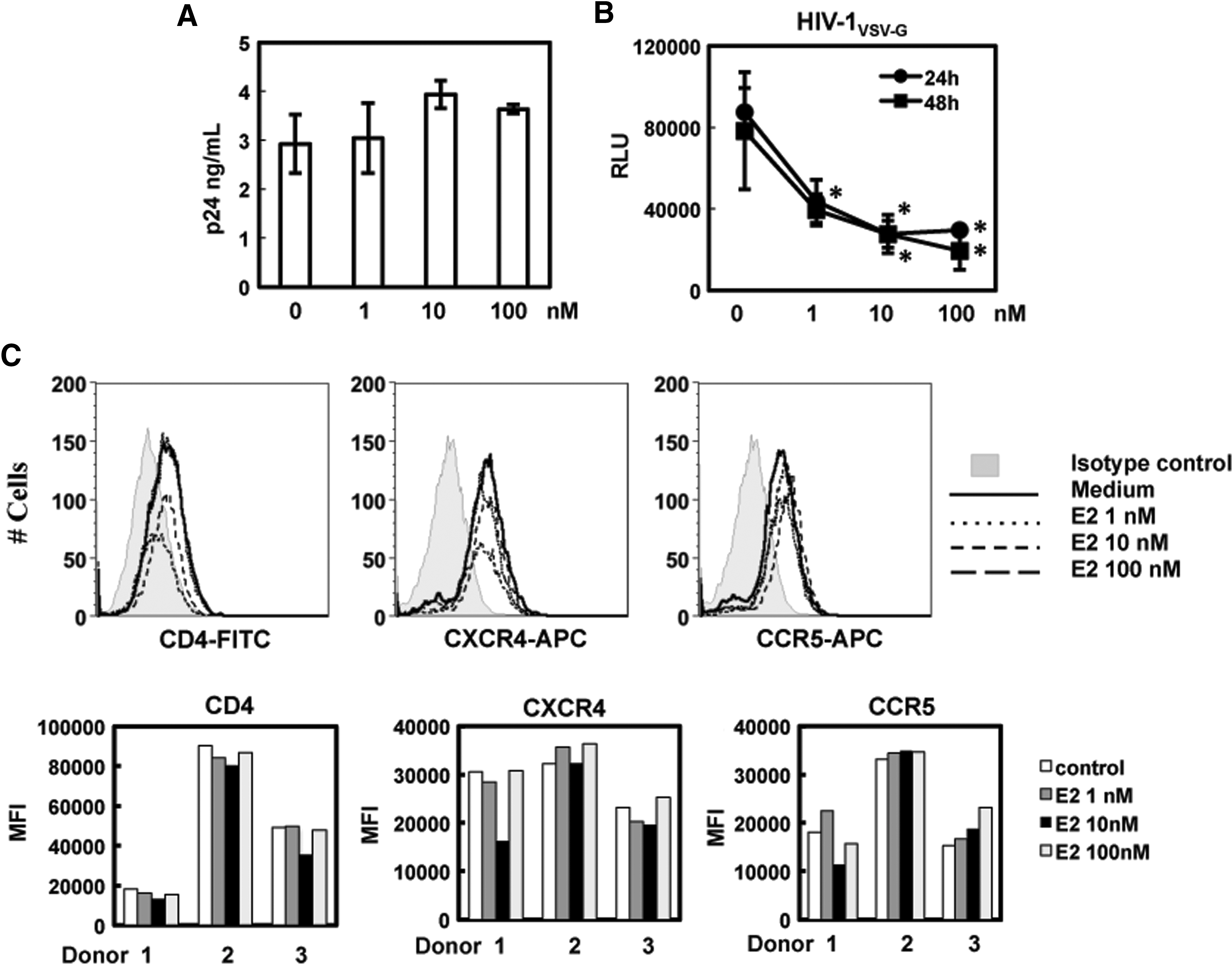
E2 does not inhibit the steps of HIV attachment and viral entry.
We then assessed the effect of E2 on the HIV reverse transcription (RT) step. The kinetics of HIV early and late RT products in MDMs with or without E2 treatment were determined by quantitative real-time PCR. Early and late RT DNA products in uninfected cells were not detectable (data not shown). In HIV-infected cells, there was no significant difference in the synthesis of early RT products between the control sample (without E2) and E2-treated MDMs (Fig. 5A). However, the synthesis of late RT products was abolished in E2-treated MDMs (Fig. 5B).

E2 blocks HIV-1 infection of MDMs at the step of late reverse transcription (RT). MDMs were treated with 10 nM E2 for 24 h followed by infection with pseudotyped HIV-1JR-FL luciferase reporter virus. Total DNA was prepared at different time points after infection, and HIV-1 RT products were determined by quantitative real-time polymerase chain reaction (PCR) using primer sets specific for early RT products
Involvement of IFNs in E2-mediated HIV infection of MDMs
To examine whether E2 induced a host restriction factor(s) in MDMs that in turn led to HIV inhibition, mRNA expression of IFNs (IFN-α, IFN-β. and IFN-γ) and known HIV host restriction factors (SAMHD1, APOBEC3G, APOBEC3F, and APOBEC3A) (2,12,20) in MDMs with or without E2 treatment was assessed by semi-quantitative real-time RT-PCR analysis. IFN-α and IFN-β were significantly induced by E2 in MDMs from several donors, whereas IFN-γ was induced only in two out of five donors (Fig. 6). Induction of host restriction factors SAMHD1, APOBEC3G, and APOBEC3F was not consistent in E2-treated MDMs. However, the induction of APOBEC3A was consistent among different donors.

E2 induces expression of interferon-alpha (IFN-α) and APOBEC3A in MDMs from different donors. MDMs were treated with E2 at 10 nM for 24 h. Total RNA was prepared, and gene expression of IFNs or specific host restriction factors was measured by semi-quantitative real-time RT-PCR. Fold of control for each gene was calculated based on ΔΔCt as described in Materials and Methods. The bars in each panel represent the fold changes in E2-treated MDMs from different donors. The gray line indicates no changes in the level of gene expression between treated and untreated samples.
IFN-α has been shown to block HIV infection at the step of late RT (7,13). Because it was consistently induced in E2-treated MDMs from many donors, we examined the involvement of IFN-α in E2-mediated HIV inhibition in MDMs. MDMs were treated with E2 in the presence of medium only (no Ab), isotype control Ab, or anti-IFN-α Ab. Anti-IFN-α Ab abolished the HIV inhibitory effect of E2, whereas E2 blocked HIV infection significantly in MDMs in the presence of medium only or isotype control Ab (Fig. 7A).

IFN-α contributes to E2-mediated HIV inhibition in MDMs.
The role of IFN-α in E2-mediated HIV inhibition was further confirmed in BMDMs from IFN-α receptor deficient mice (IFNAR(−/−)). Although hCyclin T1 is known to increase HIV RNA transcription significantly in murine cells (51), it is feasible to study HIV gene expression in BMDMs in the absence of hCyclin T1 by using pseudotyped HIV-1vsv luciferase virus to bypass the requirement for HIV entry (35). Wild-type (wt) and IFNAR(−/−) BMDMs were treated with E2 for 24 h followed by infection with pseudotyped HIV-1vsv luciferase virus. In agreement with the results using neutralizing Ab, E2 exhibited HIV inhibition in wt BMDMs. However, its HIV inhibitory effect was abolished in IFNAR(−/−) BMDMs (Fig. 7B).
To examine whether the lack of E2-mediated HIV inhibition in HIV-exposed MDMs (Fig. 3) was due to interference with induction of IFNs, MDMs were exposed to HIV-1JR-FL virus for 2 h before E2 treatment. As a comparison, MDMs were also exposed to virus without HIV Env glycoproteins (Env(−)) virus for 2 h followed by E2 treatment. Gene expression of IFNs was determined by semi-quantitative RT-PCR analysis. We found that E2-mediated induction of IFNs was partially reduced (by 37%) when MDMs were exposed to Env(−) virus. However, exposure to HIV-1JR-FL completely abolished induction of IFNs by E2 (Fig. 7C).
Discussion
The immunoregulatory functions of estrogen are well documented (reviewed in (19,46)). However, the protective role of estrogen in HIV/SIV transmission has been primarily attributed to an increase in thickness of the vaginal wall (19). A recent report demonstrates E2 inhibits HIV-1BaL replication in activated PBMCs by 50% (47). We observed a moderate inhibitory effect in activated PBMCs but not in activated CD4+ T-cells using a single-cycle infection assay (Fig. 1A–C). However, our results demonstrate that macrophages are the main target for the anti-HIV effect of estrogen. Pretreatment with E2 blocked the early phase of HIV infection of primary macrophages. E2 induced gene expression of type I IFNs and myeloid cell-specific host restriction factor APOBEC3A. We also demonstrated that IFN-α contributes to E2-mediated HIV inhibition.
HIV restriction by IFN-α has been shown to occur at the step of RT in primary macrophages, and the inhibitory effect is more pronounced on the synthesis of late RT products than on synthesis of early RT products (1,7,13). IFN-α preferentially inhibits HIV replication in MDMs compared to CD4+ T-cells (1,7,13). Our results show similar profiles of HIV restriction in E2-treated macrophages. E2-mediated HIV inhibition was found primarily in primary macrophages and occurred at the step of late RT. Our studies using neutralizing Ab against IFN-α and IFNAR(−/−) mice indicated that IFN-α played a major role in E2-mediated HIV inhibition in macrophages. IFN-α is known to induce HIV host restriction factors such as APOBEC family members and SAMHD1, which can act on early steps in the HIV life cycle (reviewed in (29,54)). Although the contribution of APOBEC3G to IFN-α-mediated HIV inhibition in MDMs has not been consistent (7,29,39), we did not observe induction of most host restriction factors (except APOBEC3A) in E2-treated MDMs among different donors. We speculate that the lack of consistent induction of APOBEC3G, APOBEC3F, or SAMHD1 in E2-treated MDMs may be because E2 treatment induces lower concentrations of IFN-α than are commonly used for exogenous stimulation (10 ng/mL). Interestingly, induction of APOBEC3A was associated with IFN-α induction and was consistently observed in E2-treated MDM from several donors (Fig. 6). APOBEC3A can be induced by IFN-α and inhibits ethe early phase of HIV infection in cells of myeloid origins (2). Thus, APOBEC3A may in part contribute to E2-mediated HIV inhibition through IFN-α induction.
Estrogen receptor (ER) signaling is activated through the binding of estrogen to ER-α or ER-β, and it has been established that cross-talk between ER signaling and the NF-κB pathway suppresses NF-κB-mediated inflammation (3,5). While the molecular mechanism of the interaction between the ER pathway and IFN signaling is not well characterized, the impact of estrogen on IFN-mediated immune responses is evident. For example, E2 suppresses IFN-γ-mediated interferon regulatory factor I (IRF-1) induction in murine lymphocytes (25), and suppresses IFN-γ-mediated interferon-induced protein of 10 kDa (IP10) induction in human keratinocytes (22). Additionally, pretreatment of monocyte-derived dendritic cells blocks type I IFN production induced by RNA viruses (11). In contrast to suppression of IFN-mediated gene induction, E2 increases IFN-γ production by activated lymphocytes from E2-treated ovariectomized mice (23). In this study, we demonstrated that E2 induces type I IFNs, and that IFN-α contributes to HIV restriction in human MDMs. Similarly, E2 induces production of IFN-α by murine plasmacytoid dendritic cells (pDCs), the major IFN-α producing immune cells, and enhances the levels of IFN-α pDCs in response to CpG, a TLR9 agonist, which modulates immune responses by pDCs (26). Although the effect of E2 on IFN responses may depend on the specific target cell and the experimental system, understanding the molecular mechanism of the interaction between ER and IFN pathways may allow us to develop specific anti-HIV inhibitors that will avoid pleiotropic effects of E2 and IFN-α.
In contrast to TLR4-mediated suppression of HIV proviral DNA activation through IFN-β in MDMs (27), E2 did not exert a significant inhibitory effect when MDMs were exposed to HIV prior to E2 treatment. Since we later discovered that E2-mediated HIV restriction occurs at the step of RT, HIV may have completed the step of RT before E2 induced sufficient amounts of IFNs to suppress HIV infection. Additionally, HIV may interfere with E2-mediated IFN induction, resulting in the lack of protection in MDMs with infection by replication competent viruses (Fig. 3C). To test this possibility, we examined the effect of E2 on IFN production in HIV-exposed MDMs, and found that induction of IFNs was abolished in HIV-exposed MDMs (Fig. 7C). We also found that HIV Env(−) virus particles also exerted a moderate inhibitory effect on E2-mediated IFN induction. The molecular mechanism underlying the interference of HIV in innate immune responses appears to be cell-type specific. HIV causes significant IRF3 degradation in transformed T-cells, although the degree of IRF3 degradation is weak in PBMCs (10). HIV accessory proteins Vpr and Vif (37) as well as Vpu (9) have been shown to be involved in IRF3 degradation. In MDDCs, the blockade of IRF3 activation rather than IRF3 degradation contributes to HIV-mediated IFN inhibition (16). Additionally, HIV Vpr but not Vif contributes to the inhibitory effect (16). Suppression of type I IFN-stimulated genes by HIV has recently been reported in primary macrophages, although the mechanism is not clear (52). We speculate that the mechanism of HIV-mediated IFN inhibition in MDMs may not be the same as in transformed cells or MDDCs because HIV particles without Env were able to suppress IFN production. The detailed mechanism is currently under investigation.
Finally, we note that our results conflict with a recent report indicating that E2 partially inhibits HIV infection of both activated CD4+ T-cells and primary macrophages (41). However, in contrast to our results, pretreatment of cells with E2 did not block pseudotyped HIV-1vsv virus. Thus, the authors concluded that E2 blocks the step of viral attachment, since expression of CCR5 was not altered in E2-treated cells. It is likely that differences in preparation of primary cells and the protocol of E2 treatment produced this discrepancy. We pretreated fully activated CD4+ T-cells with E2 versus pretreatment of CD4+ T-cells with E2 during activation (41). We treated MDMs with E2 at day 7 after differentiation, at which time the majority of MDMs expressed CD68, a differentiation marker for mature macrophages, versus treatment of MDMs with E2 at day 4 of differentiation, prior to maturation (41).
In summary, our study offers a new mechanism by which E2 protects against HIV infection through induction of IFNs. Further investigation on the interplay between HIV and innate immune responses regulated by E2 will be important for development of better strategies for HIV prevention.
Footnotes
Acknowledgments
We thank George Yap and Yungtai Lo for their help on preparation of mBMDMs and on statistical analysis, respectively. This work is supported by NIH grants AI093196, AI081559, and AI110372 (to T.L.C.). C.T. was a recipient of the Conference on Retroviruses and Opportunistic Infections (CROI) Young Investigator Award.
Author Disclosure Statement
No competing financial interests exist.