
Abstract
Hepatitis C virus (HCV) infection is a serious health problem worldwide that can lead to hepatocellular carcinoma or end-stage liver disease. Current treatment with pegylated interferon, ribavirin, and NS3/4A protease inhibitor would lead to a good prognosis in a large population of patients, but there is still no effective vaccine for HCV. HCV robustly infects hepatocytes in the liver. However, extrahepatic manifestations such as mixed cryoglobulinemia, a systemic immune complex-mediated disorder characterized by B-cell proliferation, which may evolve into overt B-cell non-Hodgkin's lymphoma, have been demonstrated. HCV-RNA is often found to be associated with peripheral blood lymphocytes, suggesting a possible interaction with peripheral blood mononuclear cells (PBMCs), especially B-cells with HCV. B-cell HCV infection was a matter of debate for a long time, and the new advance in HCV in vitro infectious systems suggest that exosome can transmit HCV genome to support “infection.” We aimed to clarify the susceptibility of primary B-cells to HCV infection, and to study its functional effect. In this article, we found that the recombinant HCV J6JFH1 strain could infect human B-cells isolated from the peripheral blood of normal volunteers by the detection of both HCV-negative-strand RNA by reverse transcription polymerase chain reaction, and NS5A protein. We also show the blocking of HCV replication by type I interferon after B-cell HCV infection. Although HCV replication in B-lymphocytes showed lower efficiency, in comparison with hepatocyte line (Huh7) cells, our results clearly demonstrate that human B-lymphocytes without other non-B-cells can actually be infected with HCV, and that this interaction leads to the induction of B-cells' innate immune response, and change the response of these cells to apoptosis.
Introduction
C
HCV is a single-stranded, positive-sense RNA virus in the Hepacivirus genus of the Flaviviridae family. Although HCV is known to infect hepatocytes in the liver and induce hepatitis in vivo, in vitro cultured primary hepatocytes barely support the HCV life cycle: only hepatoma Huh7 cells and its subclones can efficiently maintain the HCV life cycle of a very limited number of HCV strains in vitro (53).
Chronic hepatitis patients with HCV sometimes show other extrahepatic complications such as lymphoproliferative diseases (LPD), including cryoglobulinemia and B-cell malignant lymphoma, autoimmune diseases, and dermatitis (1,12,15,16). Epidemiological analysis shows that chronic HCV patients have higher rates of LPDs than non-HCV-infected populations (36,48,52). Several reports suggested that some lymphotropic HCV strains effectively infected human lymphocytes (20,47), leading to the above-mentioned abnormalities. Infection of lymphocytes with HCV has been a matter of debate for a long time. More than one decade ago, several reports described the existence of HCV-RNA in peripheral blood mononucleated cells (PBMCs) (30,40). The detection rate of HCV-RNA in PBMCs was increased if patients were infected with human immunodeficiency virus (HIV) together with HCV (44). This phenomenon indicated that immune-suppressive circumstances and/or HIV antigen might enhance the replication activity of HCV in lymphoid cells (44). Moreover, it was reported that continuous release of HCV by PBMCs was detected in HCV-infected patients, especially in HIV co-infected patients (7). In addition to HCV-HIV co-infected patients, a low level of HCV replication could be detected in peripheral lymphoid cells from HCV mono-infected patients after antiviral treatment (34,45). Moreover, it was reported that HCV persisting at low levels long after therapy-induced resolution of chronic hepatitis C remained infectious (34). This continuous viral presence could present a risk of infection reactivation.
It has been reported that HCV replication was detected in various kinds of lymphoid cells. Many reports describing the existence of HCV in B-lymphocytes and B-cell lymphoma have been published (21,25,51). Among B-lymphocytes, CD27+ memory B-lymphocytes were more resistant to apoptosis than CD27− B-lymphocytes. CD27+ B-lymphocytes were reported as a candidate subset of the HCV reservoir in chronic hepatitis C (CH-C) (38). On the other hand, others claimed that distinguishing RNA association from true HCV replication was problematic, together with multiple artifacts complicated detection and quantitation of the replicative intermediate minus strand RNA (29,31), and also the failure of retroviral (37) and lentiviral (8) pseudoparticles bearing HCV envelope glycoproteins (HCVpp) to infect primary B-cells or B-cell lines. This led to continuous debate about HCV infection into B-lymphocytes, and the riddle remained unsolved.
Using the recent progress in HCV infection systems, we intended to clarify this debate and analyze HCV infection in human lymphocytes and its functional results. Here, albeit in a lower efficiency compared to HCV infection into Huh7 cells, we report that two different strains of recombinant HCV viruses could infect primary human lymphocytes not only by the detection of HCV-RNA positive and negative strands proliferation, but also NS5A protein detection, and the detection of the activity of luciferase reporter encoded by the recombinant HCV-genome. Blocking of HCV entry using anti-CD81 antibody (Ab), and replication by IFN-α or NS3/4A protease inhibitors successfully suppressed HCV infection. We also found that HCV infection into B-lymphocytes led to the initiation of host response including apoptosis resistance.
Materials and Methods
Cells and reagents
Huh7.5.1 cells were kindly provided by Dr. Francis V Chisari (The Scripps Research Institute, La Jolla, CA). Cells were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM ; Gibco/Invitrogen, Tokyo, Japan) supplemented with 2 mM
Human peripheral blood mononuclear cells (PBMCs) were obtained from healthy volunteers by density gradient centrifugation using Ficoll Paque plus (GE-Healthcare, Waukesha, WI). CD19+ blood cells (representative of human primary B-cells) and CD19− cells (non-B-cells) were separated by MACS CD19 Beads (Milteny Biotec, Bergisch Gladbach, Germany). Purity of CD19+ B-cells was >95% after two-cycle separation. The cells were cultured in RPMI1640 (Gibco/Invitrogen) supplemented with 100 U of penicillin/mL, 100 μg of streptomycin/mL, and 10% FBS.
The following reagents were obtained as indicated: anti-CD81 Ab (BD Pharmingen, Franklin Lakes, NJ); PE anti-CD80 Ab, APC anti-CD86 Ab, and PE-labeled anti-CD19 Ab (eBioscience, San Diego, CA); recombinant IFN-α (Peprotech, Oak Park, CA); BILN2601 (Behringer, Willich, Germany); and Viaprobe 7AAD (BD Bioscience) and Annexin-V-Fluos (Roche, Mannheim, Germany).
Virus propagation
pJ6-N2X-JFH1 was kindly provided from Dr. Takaji Wakita (National Institute of Infectious Diseases, Tokyo) (2). pJc1-GLuc2A was gifted from Dr. Brett D. Lindenbach (Yale University, New Haven) (41). In vitro RNA transcription, gene transfection into Huh7.5.1 cells, and preparation of J6JFH1 and Jc1/GLuc2A viruses were performed as previously reported (53). Briefly, the HCV cDNA in plasmids were digested by XBaI and transcribed by T7 Megascript Kit (Invitrogen, Carlsbad, CA). RNA transfection into Huh7.5.1 was performed by electroporation using Gene Pulser II (Bio-Rad, Berkeley, CA) at 260 V and 950 Cap. Culture supernatant were collected on days 3, 5, 7, and 9 of postelectroporation, and concentrated with an Amicon Ultra-15 Centrifugal Filter unit (Millipore, Billerica, MA). The titer of HCVcc was checked by the immunofluoresence method using NS5A antibody when Huh7.5.1 was reinfected with these HCVcc.
Virus infection
Primary B-cells and non-B-cells were cultured with the J6JFH1 HCV strain at a multiplicity of infection (MOI)=1–3 for 3 h, and cells were harvested after four extensive washes in culture medium. On days 1–6, cells were collected, washed with 0.25% trypsin-EDTA/saline, and incubated with 0.25% trypsin-EDTA for 5 min at 37°C. Then, suspended cells were collected as a source of total RNA. In some experiments, B-cells were infected with the Jc1/GLuc2A strain at MOI=5 for 3 h. Cells were washed five times in 1× phosphate buffered saline (PBS), and cultured until day 6 for determination of viral replication as GLuc activity with BioLux Gaussia luciferase assay kits (41).
RNA purification, RT-PCR, and quantitative PCR
Total RNA was extracted by using Trizol Reagent (Invitrogen) according to the manufacturer's instructions. Using 100–400 ng of total RNA as a template, we performed RT-PCR and real-time RT-PCR as previously described (3,4). Primer sets are shown in Supplementary Table S1 and Table S2 (Supplementary Data are available online at
Real-time PCR was used for quantification of positive-strand and negative-strand HCV RNA. Total Trizol-extracted RNA was analyzed by RT-PCR with a modification of the previously described strand-specific rTth RT-PCR method (10,13). RT primers for complementary DNA synthesis of positive and negative strand HCV RNA are shown in Supplementary Table S1. Positive-strand and negative-strand HCV PCR amplifications were performed using Power SYBR Green PCR Master Mix (Applied Biosystems, Warrington, UK) with 200 nM of paired primers (Supplementary Table S1). The PCR conditions were 95°C for 10 min, followed by 40 cycles at 95°C for 15 sec and 60°C for 1 min.
Virus production and releasing assay
Primary human B-cells were infected with J6JFH1 at MOI=1. Six days postinfection, the supernatant was collected (“releasing samples”), cells were repeatedly frozen and thawed, and the supernatant was collected (“assembly samples”). Viral titers of “releasing samples” and “assembly samples” were determined with Huh7.5.1 cells using J6JFH1 virus (MOI=0.001 and 0.01) as control. Total RNA was recovered from the cells on days 2, 4, and 6, and determined with HCV-RNA to check reinfectivity.
Indirect immunofluorescence
Indirect immunofluorescence (IF) expression of HCV proteins was detected in the infected cells using rabbit IgG anti-NS5A antibody (Cl-1) (3). Goat anti-rabbit Alexa 594 (Invitrogen) was used as secondary Ab. Fluorescence detection was performed on the Zeiss LSM 510 Meta confocal microscope (Zeiss, Jena, Germany) (13).
Luciferase assay
Primary B-cells were infected with Jc1/Gluc2A by using concentrated Medium or Mock Medium (PBS-electropolated Huh7.5.1 medium). Media were collected on days 0, 2, 4, and 6 postinfection, cleared by centrifugation (16,000 g for 5 min), and mixed with 0.25 volume of Renilla 5 lysis buffer (Promega, Madison, WI) to kill HCV infectivity. GLuc activity was measured on a Berthold Centro LB 960 luminescent plate reader (Berthold Technologies, Bad Wildbad, Germany) with each 20 μL sample injected with 50 μL BilLux Gaussia Luciferase Assay reagent (New England Biolabs, Ipswich, MA), integrated over 1 sec.
Cell survival assay
Apoptosis assay: Primary B cells were infected with J6JFH1 virus. Cells were collected 48 h after infection, stained by 7AAD Cell Viability assay kit and Annexin V, and analyzed by FACS Calibur (BD) (13).
ATP assay
Primary B-cells were infected with J6JFH1 virus or Mock concentrated medium. Cells were resuspended and cultured at Lumine plate (Berthold Technologies) postinfection. ATP activities were determined 72 h later using CellTiter-Glo® Luminescent Cell Viability Assay (Promega) according to the manufacturer's protocol.
miRNA detection
Total RNA was extracted by using Qiazol Reagent (Invitrogen). These RNA was purificated and reverse transcripted to cDNA by using the miScript II RT Kit. Synthesized cDNA was used to determine the expression levels of miR-122 (24). Total miRNA was prepared by using Qiazol and miScript II RT kit (Invitrogen), and miR-122 expression was determined by using miScript SYBR Green PCR Kit and miScript Primer Assay (Invitrogen) according to the manufacturer's protocol. U6 small nuclear RNA was used as an internal control.
Results
J6JFH1 infects and replicates in primary B-cells
To address HCV infectivity into primary B-cells, PBMC were isolated from the blood of healthy volunteers and were sorted into CD19+ cells (primary B-lymphocytes) and CD19− cells (non-B-cells). Their purities were >95%. These cells were then incubated with the J6JFH1 HCV. Total RNA was collected on days 2, 4, and 6. The Huh7.5.1 strain was used as positive control. Both Huh7.5.1 and primary B-cells, but not non-B-cells, showed an increase in intracellular HCV-RNA titer, albeit primary B-cells showed lower efficiency than Huh7.5.1 (Fig. 1A). We adjusted the HCV-RNA values using GAPDH as an internal control (Fig. 1B). To confirm J6JFH1 replication in primary B-cells using IF, we also measured the expression of HCV-NS5A, which is a nonstructural protein produced only by the virus secondary to replication. Although the expression was far lower than Huh7.5.1 cells, we managed to detect the NS5A expression in J6JFH1 infected primary B-cells (Fig. 1C).
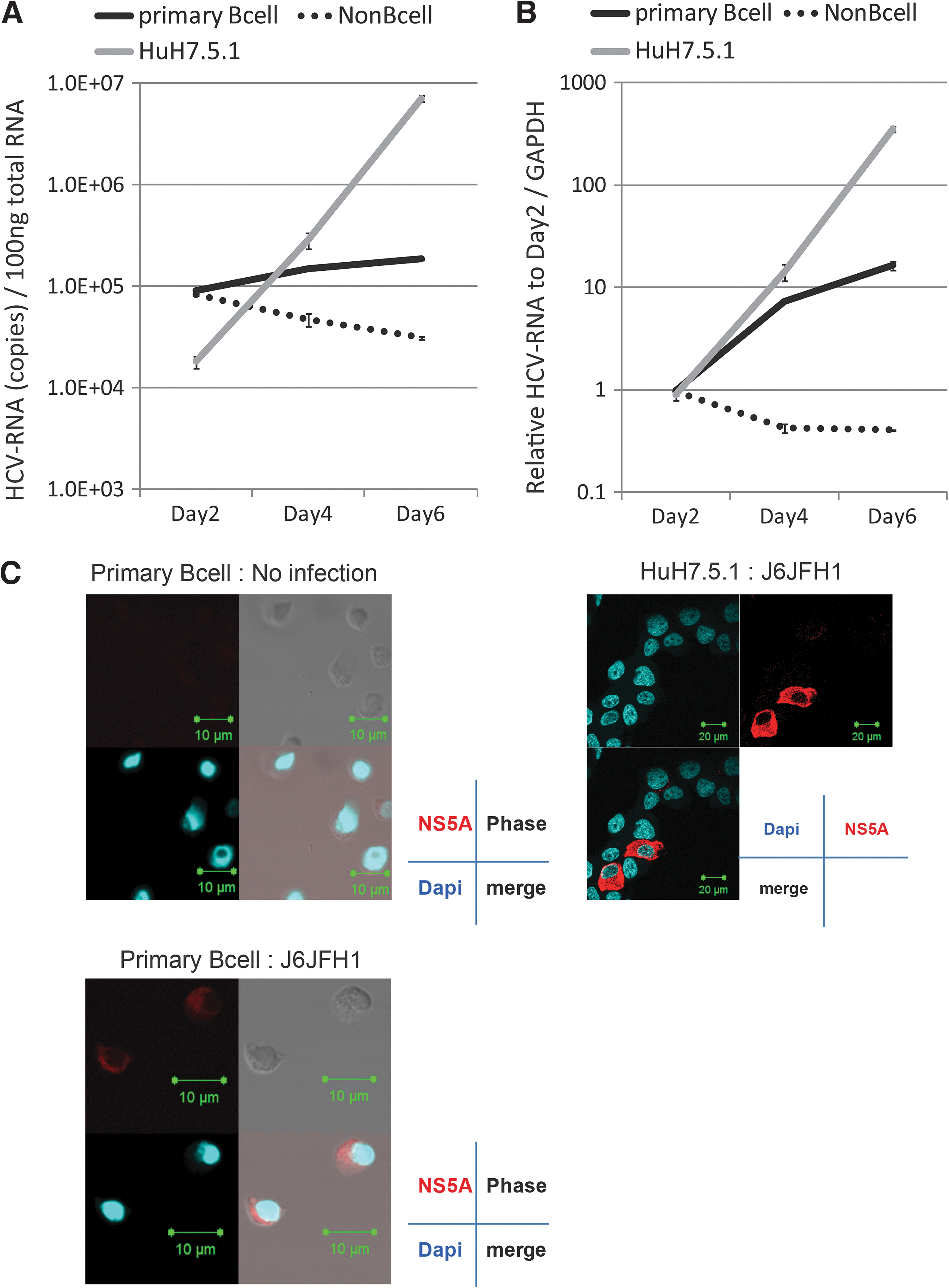
J6JFH1 infects human peripheral blood B-cells. Human B-cells (CD19+ cells) and non-B-cells (CD19− cells) were separated by MACS as described in Materials and Methods. Primary B-cells, non-B-cells, and Huh7.5.1 cells were infected with J6JFH1 at MOI=1 for 3 h. After infection, cells were washed twice with culture medium and continued culture. On days 2, 4, and 6, total RNA was collected and HCV-derived RNA was determined by reverse transcription polymerase chain reaction (RT-PCR). GAPDH was used as internal control.
We examined what kinds of HCV-entry receptors human primary B-cells expressed in our setting. Human CD81, SRB1, and NPC1L1 were expressed, but not the tight junction proteins claudin1 and occludin in mRNA levels (Supplementary Fig. S1). We could not detect miR122 in primary B-cells (Supplementary Fig. S2), expression of which makes the cells permissive to HCV (24). Human CD81 is a primary entry receptor for HCV in hepatocytes (42). Blocking human CD81 by its specific Ab resulted in blockage of HCV infection into primary B-cells, as shown by the suppression of HCV-RNA titer (Fig. 2), suggesting that HCVcc particles enter B-cells also using CD81 receptor. HCV-RNA titer was not suppressed by non-specific Ab (data not shown).
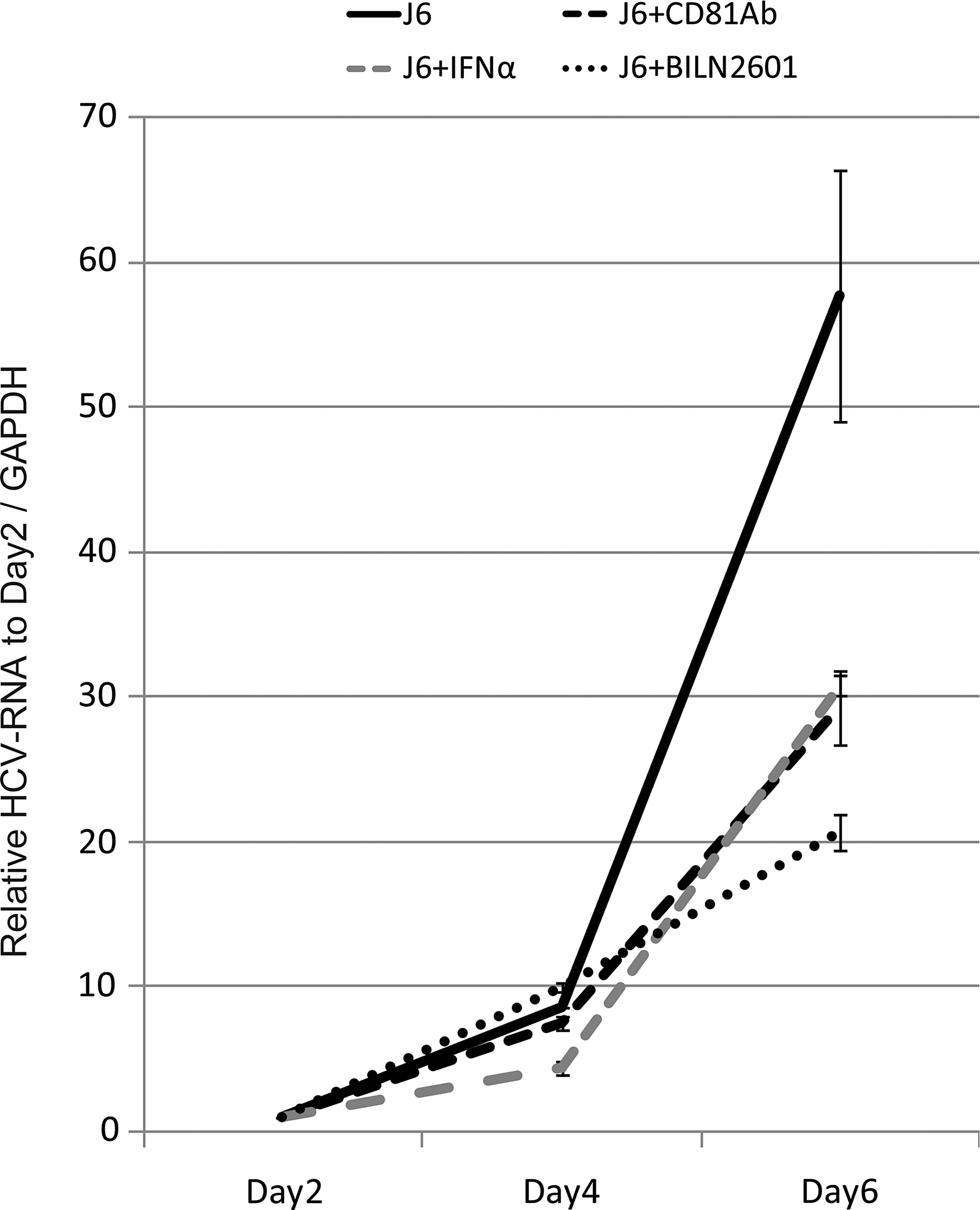
J6JFH1 B-cell infection is blocked by anti-CD81 Ab, IFN-α, or an NS3/4A inhibitor. Anti-CD81 neutralizing Ab (20 μg/mL) was added to the B-cell culture 1 h before infection. Otherwise, recombinant IFN-α rhIFN-α, 200 IU/mL) or BLIN2601 (250 nM, which is IC75; see Supplementary Fig. S3) was added 1 h after infection. On days 2, 4, and 6, total RNA was extracted, and HCV-RNA was determined by RT-PCR. The values were adjusted by GAPDH.
We then examined the effect of the different drugs used to suppress HCV replication (recombinant human IFN, and HCV protease inhibitor, BILN2601). Inhibition of HCV-RNA replication was observed when B-cells were treated with rhIFN-α or BILN2601 (Fig. 2) after infection. BILN2601 showed efficient inhibitory effect on replication of HCV RNA in Huh7.5.1 cells (Supplementary Fig. S3). As control studies, we confirmed that the production of HCV RNA was reduced in Huh7.5.1 cells by CD81 Ab, IFN-α, or BLIN2601 (Supplementary Fig. S4). In both Huh 7.5.1 and B-cells, BLIN2601 most effectively block HCV replication. These data reinforce that HCV is actually replicating in primary B-cells, and that activation of innate immunity by IFN treatment or blocking the NS3/4A protease function is a critical factor in blocking HCV replication in primary B-cells. These data suggest that our system can be used for screening the function of different inhibitors on HCV replication in B-cells.
HCV negative-strand RNA detected in human B-cells
To confirm HCV replication in primary B-cells further, we tested for an increase of negative-strand HCV-RNA after infection, since the negative-strand RNA is not yielded if HCV particles or RNA just adhere to the cell surface of human primary B-cells without internalization (9,14,19,35, 42,43). We measured the synthesis of plus-strand and minus-strand HCV-RNA separately using strand-specific RT primers and rTth polymerase as previously described (4). The titer increase of minus-strand HCV-RNA indicates HCV-RNA replication. As shown in Figure 3, both minus- and plus-strand HCV-RNA increased time dependently in primary B-cells, and both types of RNA concomitantly decreased in non-B-cells (Fig. 3A and B). Plus- and minus-strand RNA were exponentially increased in Huh7.5.1 cells infected with J6JFH1 (Fig. 3C). These results indicated that primary human B-cells supported J6JFH1 infection and replication, although viral replication levels in B-cells were modest compared with those in Huh7.5.1 cells. These results may reflect the fact that the NS5A protein is difficult to detect in infected B-cells using IF assay.
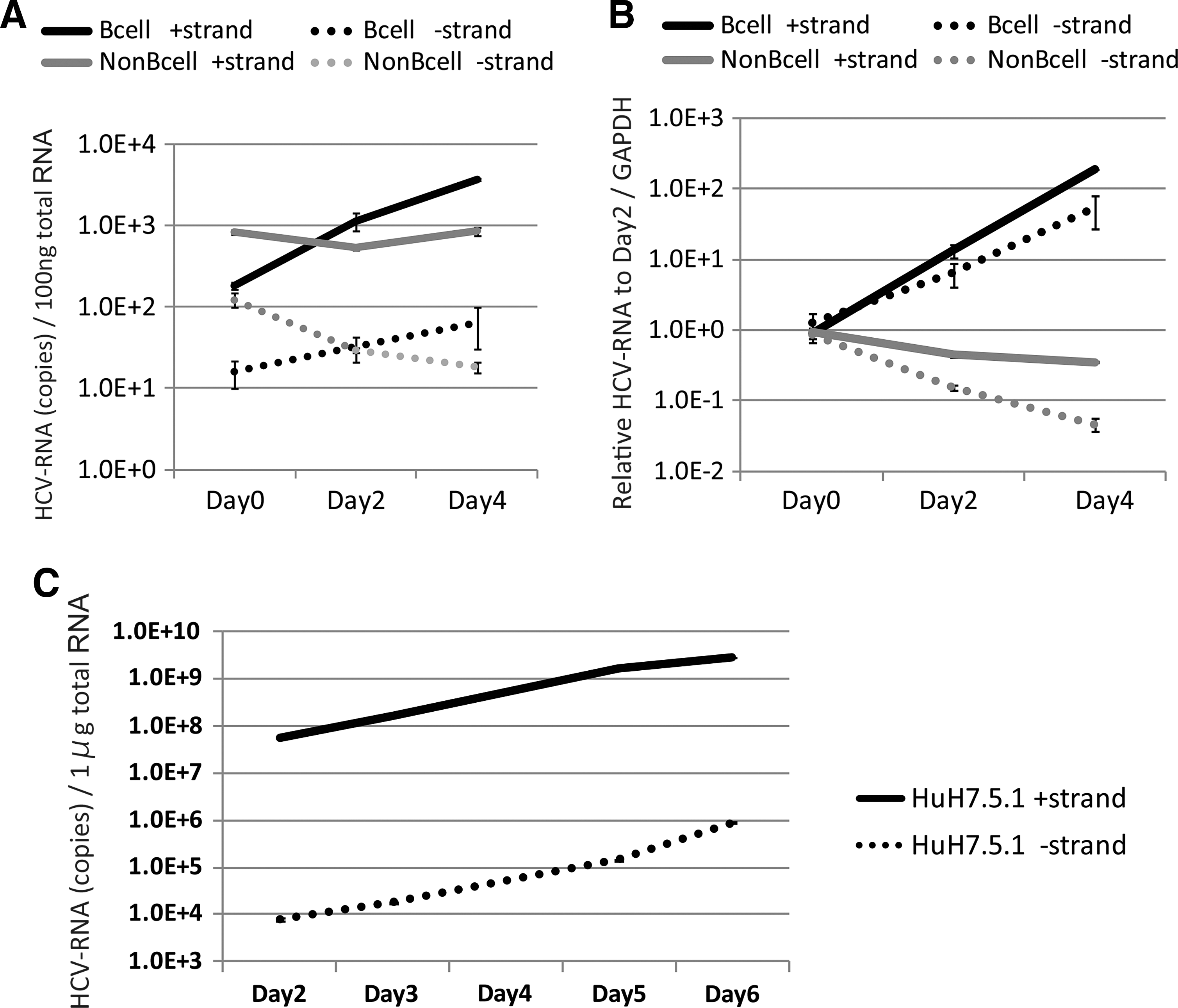
HCV negative strand RNA is detected in human B-cells. By using rTth methods, HCV strand-specific RNA was determined in J6JFH1-infected human B-cells.
B-cells can be infected with different HCV strains
We next used the Jc1/GLuc2A strain to investigate whether different HCV strains infect primary B-cells. Primary B-cells, non-B-cells (data not shown), and Huh7.5.1 cells were infected with the Jc1/GLuc2A strain. After five washes, supernatant was collected (day 0 samples). On days 2, 4, and 6, medium was collected. Luciferase activity was determined for all samples by luminescence (GLuc). GLuc activity and detection of RNA increased exponentially in Huh7.5.1 cells infected with the Jc1/GLuc2A strain (Fig. 4A). GLuc activity on day 4 to day 6 increased more in primary B-cells than in non-B-cells (Fig. 4B). These results suggest that HCV replication is substantial, but low in the HCV line Jc1/GLuc2A.
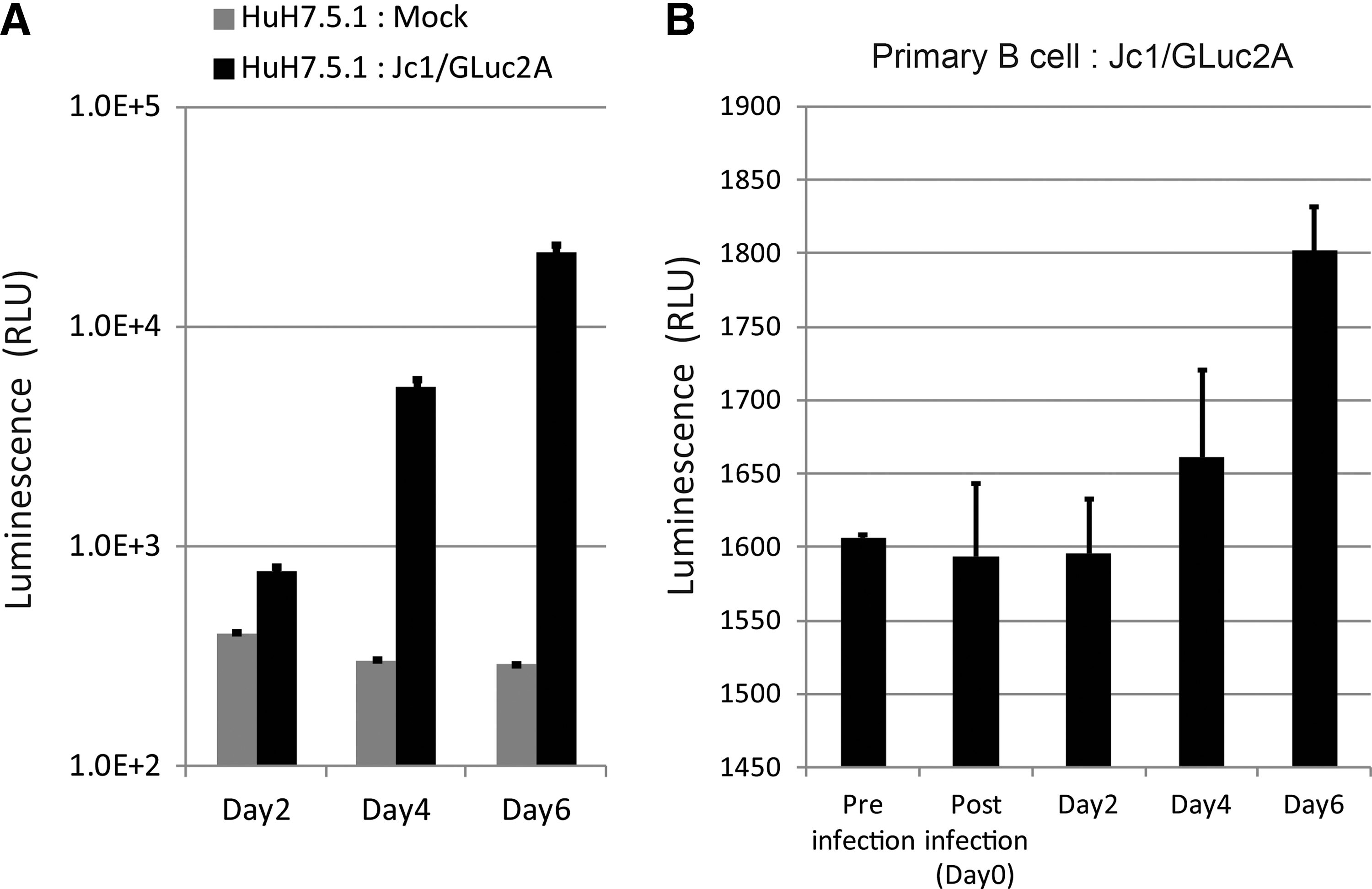
Jc1/GLuc2A strain infects human B-cells with an increase of Gluc activity. Human B-cells and Huh7.5.1 cells were infected with the JC1/GLuc2A strain that contains secretory luciferase derived from Gaussia (GLuc) at MOI=5. Huh7.5.1 cells were used as control. GLuc activity was increased as time cultured. The GLuc activity was saturated in Huh7.5.1
B-cells neither produce nor release detectable level of HCV infectious particles
We collected supernatants of J6JFH1-infected primary human B-cells to measure productive infection in B-cells. The supernatant was then added to culture of Huh7.5.1 cells, and we compared infection with control Huh7.5.1 cells, whose cells were infected with a low MOI (0.01 and 0.001) of J6JFH1 collected from media of the infected Huh7.5.1 cells. HCV-RNA titer in the Huh7.5.1 titrating cells was decreased over time after co-culture with B-cell supernatants obtained from either “releasing samples” “assembly samples.” In contrast, HCV-RNA titers were slightly increased over time in the Huh7.5.1 titrating cells that had been infected with medium collected from low MOI-J6JFH1-infected Huh7.5.1 cells (Fig. 5). These results indicated that primary human B-cells were infected with J6JFH1 but failed to assemble or produce particles into the supernatant.
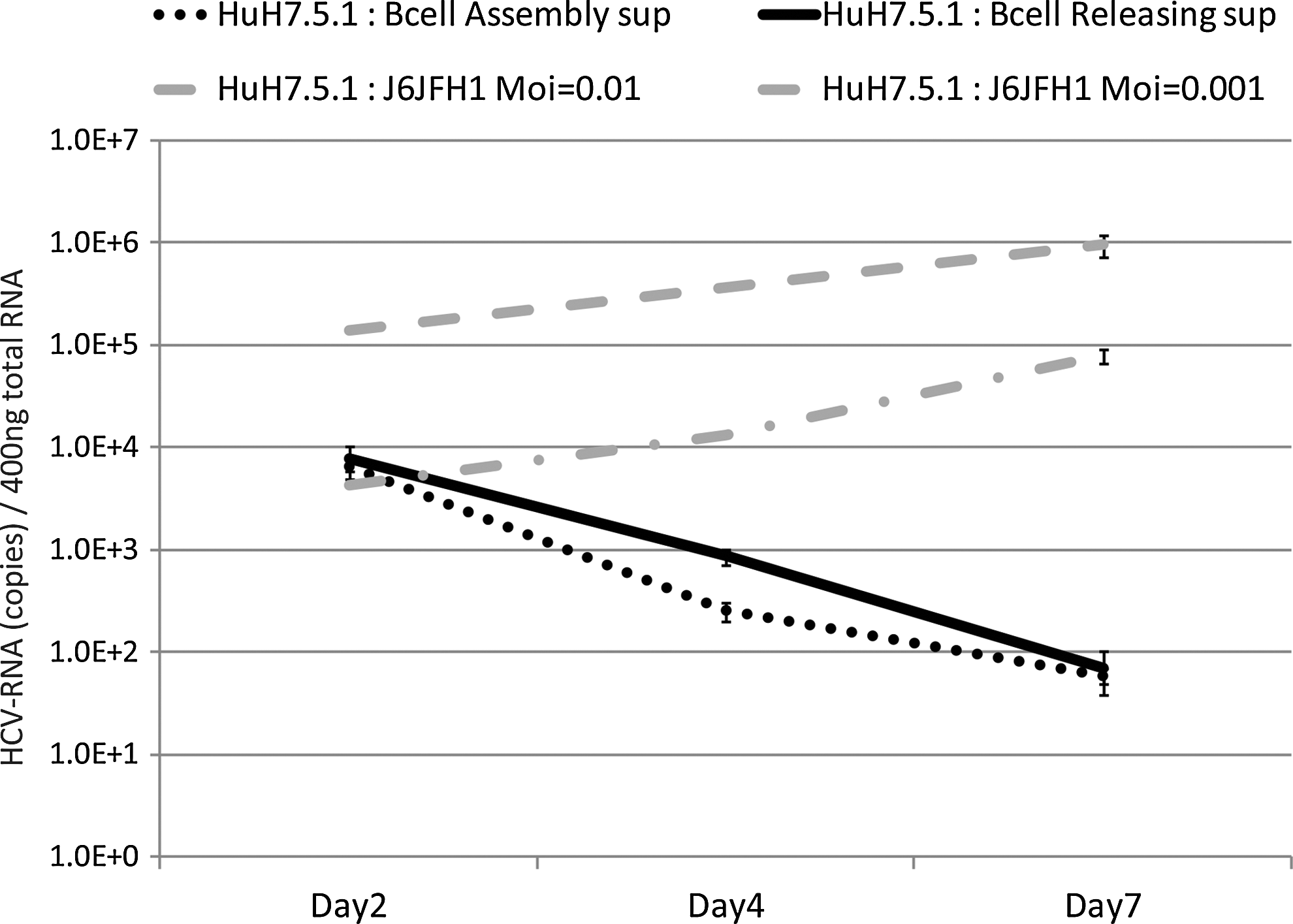
B-cells infected with J6JFH1 fail to produce virus particles. Human B-cells were infected with J6JFH1 for 3 h, washed twice with phosphate buffered saline (PBS), and cultured. Six days after infection, the supernatant was collected (“releasing samples”). Cells were periodically frozen and thawed five times, and the supernatant was collected (“assembly samples”). For evaluation of the infectious virions, Huh7.5.1 cells were treated with these “releasing samples” or “assembly samples.” Similarly, Huh7.5.1 cells were treated with J6JFH1 at low MOI (MOI=0.01 and 0.001) in parallel. After the treatment, cells were washed and cultured. On days 2, 4, and 7, cells were harvested to collect HCV-RNA. Total RNA was extracted from each samples, and HCV-RNA was determined by RT-PCR methods.
Host response to HCV infection into primary B-cells
Next, we determined whether B-cell activation was induced in HCV-infected B-cells that survived under HCV infection. We measured induction of CD80 and CD86 as B-cell activation markers. After 2–3 days of infection, the CD80/86 levels on B-cells treated with J6JFH1 were compared with those treated with medium from mock-infected cells (concentrated Huh7.5.1 medium) by FACS analysis (Fig. 6A). We found that CD80/86 were upregulated in infected cells compared to mock-infected cells.
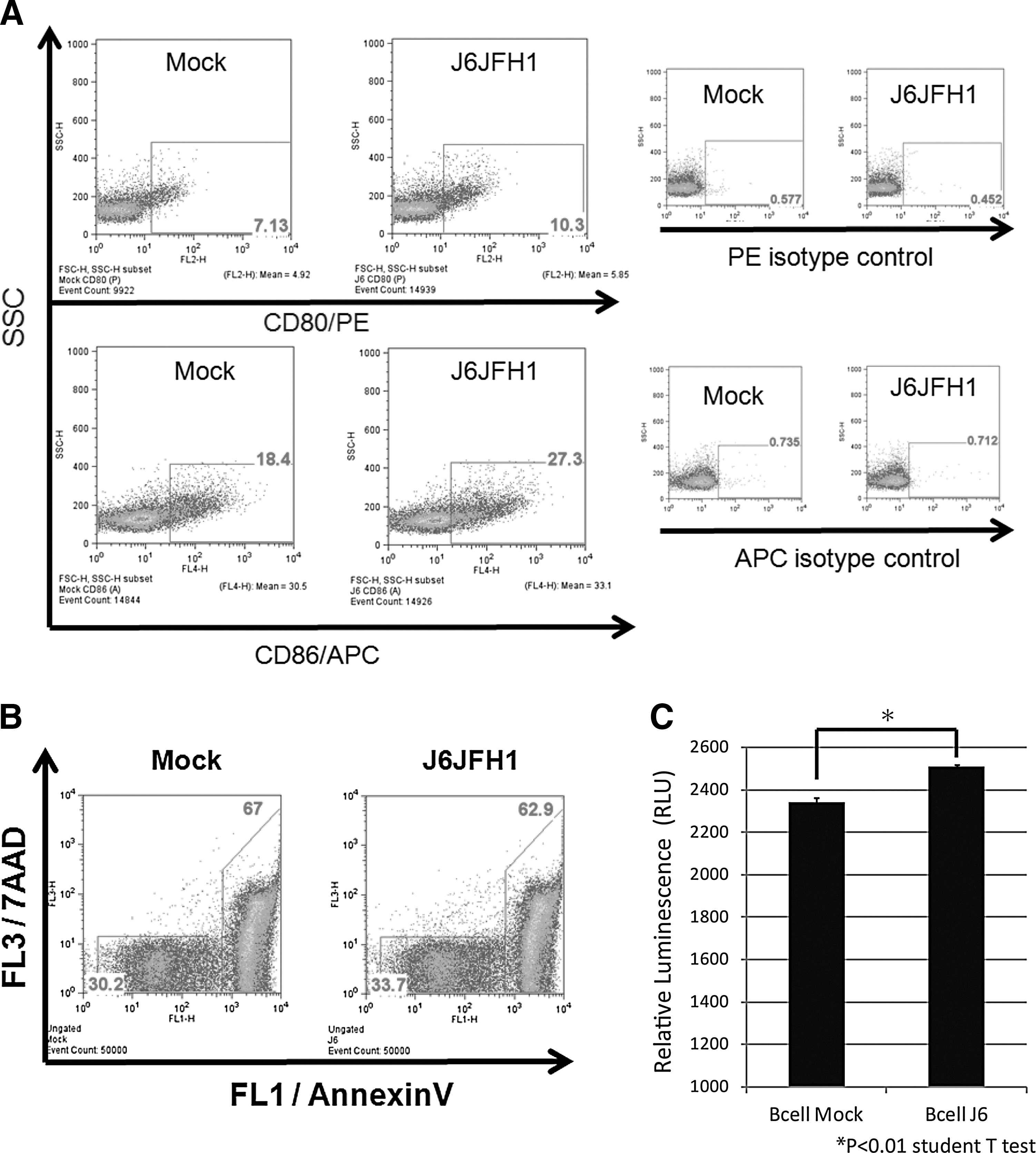
J6JFH1 infection activates B-cells and protects the cells from apoptosis. Human B-cells were infected with J6JFH1 at MOI=1 for 3 h, washed twice with PBS, and cultured. Two days after inoculation, cells were washed and suspended with FACS buffer.
Since B-cell lymphoma is a known complication of chronic HCV infection (20,36) and acquiring apoptotic resistance is essential for the development of cancer (21,51,38), we measured the ability of B-cells to escape apoptosis after HCV infection. B-cell apoptosis spontaneously occurs during culture at 37°C. The percent of apoptosis of primary B-cells was decreased in FACS analysis using 7AAD viaprobe + annexinV (Fig. 6B) and ATP assays postinfection (Fig. 6C). These results suggest that primary B-cells are protected from apoptosis by infection with HCVcc. It has been reported that B-cells were vulnerable to apoptotic cell death at various stages of peripheral differentiation and during signal responses (18). Thus, the results infer that HCV stimulation interferes with B-cell apoptotic signal in human B-cells.
Discussion
We show evidence suggesting that human peripheral B-cells can be infected with HCV strains. Establishment of J6JFH1 infection was evaluated by minus-strand PCR amplification, production of core and NS5A proteins, and protection from apoptosis. An increase in HCV RNA in B-cells was inhibited by an exogenously added antibody against CD81 that blocked HCV receptor function. Furthermore, blocking HCV replication in B-cells by type I IFN and NS3/4A protease inhibitor confirmed the presence of HCV infection/replication in human B-cells. The results were corroborated with another HCV strain, Jc1/GLuc2A. Although we failed to establish an EBV-transformed B-cell line to reproduce HCV infection of B-cells, peripheral blood B-cells were infected with J6JFH1 in 12 independent experiments.
One of the well-known complications of chronic HCV infection is LPD, including cryoglobulinemia and B-cell malignant lymphoma, indicating the involvement of B-cells in the course of the disease (1,12,15,16). However, many reports describing the existence of the HCV genome in B-cells and lymphomas (21,25,51) and HCV replication in B-cells have been controversial due to multiple artifacts complicated in detection and quantitation of the replicative intermediate minus strand RNA (29,31). This has led to a continuous debate about HCV infection in B-lymphocytes.
HCV entry into B-cells has also been previously reported to be absent because retroviral (37) and lentiviral (8) pseudoparticles bearing HCV envelope glycoproteins (HCVpp) did not infect primary B-cells or B-cell lines. In our study, while we succeeded in infecting Huh7.5.1 cells efficiently with retroviral pseudoparticles for expressing both HCV E1/E2 and the control VSV-G, we failed to establish the same infection in B-cells, suggesting that the block of pseudoparticle entry into B-cells is not related to HCV glycoproteins alone.
Total PBMCs reportedly facilitate HCV attachment but not internalization (42), so HCV infection of B-cells is abrogated in total PBMCs (35). The cause of HCV absorption is unclear, but incomplete sets of HCV receptors in non-B PBMC cells permit attachment of HCV without internalization. B-cells possess CD81, SRBI, LDL-R, and NPC1L1. Because B-cells are not adherent cells, they do not express claudin 1 and occludin, which forms a receptor complex for HCV (9,14,19,43). Claudin 1 and occludin are components of tight junctions and serve as HCV receptors in human hepatocytes. In infection studies using cells expressing these proteins, however, claudin 1 and occludin only upgrade infection efficacy and are dispensable to infection (5), although CD81 is essential for establishment of infection (42). Lack of claudin 1 and occludin or miR122 might be a cause of the low HCV infection efficiency observed in human B-cells. Function blocking of CD81 by its specific antibody suppressed HCV infection in primary B-lymphocytes, which imply that HCV entry into primary B-lymphocyte is dependent on the direct interaction phenomenon between HCV virus particles and CD81 receptor and is not mediated by other nonspecific (CD81 independent) pathways such as exosomal transfer of HCV from Huh7 cells to nonhepatic cells, such as dendritic cells (46).
Previous report using in vitro prepared recombinant HCV JFH1 particles (HCVcc) failed to establish HCV infection in B-lymphocyte cell lines (39). While HCV is known to infect human hepatocytes in vivo leading to chronic viral hepatitis, in the in vitro conditions, only the combination between Huh7 cells and its derived clones supported robust replication and infection with only JFH1 or its derived chimeras (5). Neither hepatocyte cell lines including primary hepatocytes nor other HCV strains could reproduce HCV infection efficiently in vitro (5). These data suggest that the clonal selection of HCV quasispecies by hepatoma Huh7 cells is essential for this robust infection in vitro. The situation would be similar to the JFH1 story in B-cell HCV infection.
B-cell apoptosis spontaneously occurs during culture at 37°C. We found that B-cell apoptosis was blocked by J6JFH1 infection, as reported previously using Raji cells (11). B-cell apoptosis usually occurs secondary to viral infection, but HCV is particular since apoptotic signaling interferes with infection, leading to protection from cell death. However, B-cell survival was not due to primary infection, because the percent of cells circumventing apoptosis was usually higher than cells infected with HCV. We could not define the pathways that participated in apoptosis regulation by HCV, although a previous report (11) suggested that E2-CD81 engagement was related to B-lymphocyte disorders and weak neutralizing antibody response in HCV patients. Since B-cell lymphoma is a known complication of chronic HCV infection (27), the inability of infected cells to undergo apoptosis can be associated with the development of cancer (28,33,49). In this context, B-cell lymphoma often occurs in mice with Cre-initiated HCV transgenes (26). It is notable that anti-apoptotic effect of HCV core gene was reported in genotype 3a in Huh7 cells (23) and, here, genotype 2a in B-cells. In another report (51), HCV strains established from B-cell lymphoma persistently infected with HCV were genotype 2b. B-cell HCV infection might not be linked to some specific genotypes of HCV.
We believe that our report shows that human primary B-cells can be infected in vitro with HCV, and that this infection is dependent on HCV particles binding with its receptor CD81 and is not nonspecific entry (e.g., exosomal mediated). We also show that this infection could be blocked with antibodies interfering with this binding, or with drugs that suppress HCV replication. Although no virion was generated from B-cells in HCV infection, it is still likely that B-cells serve as a temporal reservoir of HCV in the blood circulation. If B-cells permit HCV infection, RNA sensors RIG-I and MDA5 in B-cells might recognize HCV RNA and evoke intracellular signaling, including by transcription factors NF-κB and IRF-3/7 (5). Activation of the cytokine network is triggered in human B-cells in response to HCV RNA. In fact, host factors liberated by HCV-infecting B-cells have been previously reported in HCV patients (1,12,15,16,52). Although patients' outcomes would be more than we can be predicted from our results, this system would actually benefit the future study on B-cell–virus interaction.
Footnotes
Acknowledgments
We are grateful to Drs. Frank Chisari (Scripps Research Institute, San Diego, CA) for the Huh7.5.1 cells, Takaji Wakita (National Institute of Infectious Diseases, Tokyo) for supplying the J6JFH1 plasmid, and Brett Lindenbach (Yale University, New Haven, CT) for providing us with the pJc1-GLuc2A HCV strain.
This work was supported in part by Grants-in-Aid from the Ministry of Education, Science, and Culture (Specified Project for “Carcinogenic Spiral”) and the Ministry of Health, Labor, and Welfare of Japan, and by the Program of Founding Research Centers for Emerging and Reemerging Infectious Diseases, MEXT. Financial support by the Takeda Science Foundation, the Yasuda Cancer Research Foundation, and the Iskra Foundation are gratefully acknowledged.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.