
Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) glycoprotein 5 (GP5) is the most abundant envelope glycoprotein and a key target for neutralizing antibodies. Previous studies have demonstrated that the native GP5 glycoprotein is poorly immunogenic and not able to induce robust protective responses, probably due to the presence of a non-neutralizing decoy epitope and shielding N-linked glycans close to its neutralizing epitope. In the present report, two Fc tagged GP5 proteins (GP5-Fc containing a truncated GP5 with the deletion of its signal peptide and transmembrane regions, and GP5N-Fc containing only the ectodomain of GP5) were designed based on the sequences of a highly pathogenic PRRSV strain and produced using a baculovirus/insect cell expression system. Immunization studies showed that both GP5-Fc and GP5N-Fc elicited strong serum responses in the absence of adjuvant to the native GP5 present on the surface of purified PRRSV virons. Although GP5N-Fc failed in inducing significant titer of neutralizing antibodies in mice, GP5-Fc was shown as an effective inducer of neutralizing antibodies specific to PRRSV. Our results suggest that the modified GP5 glycoprotein with Fc tag has the potential to be a candidate for the future development of new generation vaccine against PRRSV infection.
Introduction
P
The PRRSV genome is approximately 15 kb, and it comprises at least nine open reading frames (ORFs), with the ORF1a/b encoding viral replicase polyproteins and the ORF2-7 encoding structural proteins (19,20). Recently, a novel structural protein ORF5a has been discovered in all arteriviruses (8,12), but the function of this protein is still unknown, and its protein immunization failed to induce protective immunity against PRRSV infection (27).
ORF5-encoded GP5, which is involved in the recognition of the host cell receptor sialoadhesin on macrophages (29), is a major envelope glycoprotein, as well as a key target for PRRSV neutralizing antibodies. Previous studies have shown that antibodies to GP5 are responsible for virus neutralization in natural PRRSV infection (9), and monoclonal antibodies (mAbs) of GP5 can neutralize homologous strains of PRRSV infection in vitro (23,31).
GP5 is about 25 kDa in molecular weight, comprising a hydrophilic N-terminal signal peptide, a short extracellular domain (ectodomain) with several putative N-glycosylation sites, three hydrophobic transmembrane helices, and a large C-terminal intracellular domain (endodomain) (25). Neutralizing epitopes have been defined as linear peptides in the amino acids 29–35 of the European representative strain Lelystad virus (33), and amino acids 37–45 or 36–52 of the North American strain VR-2332 (22,24). Although peptides resembling the GP5 neutralizing epitope failed to stimulate protective antibodies (16), a number of reports showed that immunization of animals with GP5 derived from mammalian (6), insect (25), yeast (37), or bacterial expression systems (26) could elicit neutralizing antibodies and provide protection against PRRSV infection.
IgG Fc fragment can recognize Fcγ receptors on antigen presenting cells (APC). It has been suggested that fusion with Fc-tag could facilitate the uptake of antigens and enhance their immunogenicity, and this strategy has been utilized in the design of adjuvant-free vaccines to prevent cancer (10) and virus infections such as HIV-1 (3,35,36) and influenza (17).
The aim of this study was to modify GP5 to enhance its immunogenicity for future development of a new generation of PRRSV vaccine. To this end, a truncated GP5 gene with the deletion of its signal peptide and transmembrane regions was constructed with the strategy described by Ren et al. (26), and the truncated GP5 protein was expressed and purified from a prokaryotic system. Two more Fc-tagged proteins, with GP5N-Fc containing only the ectodomain and GP5-Fc containing both the ectodomain and endodomain of the truncated GP5, were then produced using baculovirus/insect cell expression system. The Fc fusion proteins were used to immunize mice in parallel with the GP5 protein produced in Escherichia coli. The immunogenicity of these proteins were evaluated by antibody titration and neutralization assays.
Materials and Methods
Construction of expression vectors
GP5 gene with the deletion of the signal peptide and transmembrane regions was constructed according to the strategy described by Ren et al., and the extracellular and intracellular domains were linked with three repeats of GGGGS amino acid sequences (GGGGS×3 linker) (26). The truncated gene, which was based on the GP5 sequences of a highly pathogenic PRRSV strain TA-12 (GenBank accession number HQ416720.1) and flanked by two SfiI, was synthesized by Shanghai Xuguan Biotechnological Development Co. Ltd.
Sequences of primers used in this study are listed in Table 1. To generate the construct of pET-28a-GP5, the GP5 gene was amplified from the synthesized gene by polymerase chain reaction (PCR), using primers BspGP5-F and XhoGP5-R. The PCR product was digested with BspHI and XhoI, and cloned into pET-28a (Novagen) between the NcoI and XhoI sites. To make the Fc tagged construct of pAcGP5Fc, the GP5 gene containing both the ecto- and endo- domains was cut from the synthesized template using SfiI, and cloned into the SfiI sites of pAcFc vector (3). This vector contains a polyhedrin promoter for the gene expression in baculovirus infected insect cells and gp64 signal peptide sequences for the secretion of the produced protein. For pAcGP5NFc, the gene fragment containing only the ectodomain (GP5N) was amplified using primers SfiGP5-F and SfiGP5-R, and also cloned into the SfiI sites of pAcFc vector.
To make GST fusion proteins, GP5N gene was amplified using primers BamGP5N-F and EcoGP5N-R, and GP5C was amplified using BamGP5C-F and EcoGP5C-R. The PCR products were digested with BamHI and EcoRI, and then cloned into pGEX-2T to generate pGEX-2T-GP5C and pGEX-2T-GP5N constructs.
Expression and purification of the proteins
The bacterial expression plasmids pET-28a-GP5, pGEX-2T-GP5C, and pGEX-2T-GP5N were transformed into E. coli BL21(DE3) strain. Protein expression was induced by 1 mM isopropyl-β-
The transfer vectors pAcGP5Fc and pAcGP5NFc were co-transfected with linearized Bacmid DNA (38) into Spodoptera frugiperda (Sf9) cells (Invitrogen) to make recombinant baculoviruses expressing the Fc-tagged proteins. Sf9 cells were maintained at 27°C in SFX-INSECT medium (Thermo Scientific HyClone) with 1% fetal bovine serum (Thermo Scientific HyClone). The Fc fusion proteins secreted into the cell media were harvested 3 days postinfection of the recombinant baculoviruses, and purified by affinity chromatography using Protein A Agarose (Beijing CoWin Biotech).
Vaccination of mice
Female inbred SPF BALB/c mice aged 3–4 weeks were purchased from the Animal Center of Xi'an Jiaotong University and allowed free access to food and water. Three groups of mice (five mice per group) were subcutaneously immunized, 10 μg per injection, with purified GP5, GP5-Fc, or GP5N-Fc. Four immunizations were made at 2 week intervals. As the Fc tag has adjuvant activity, GP5-Fc and GP5N-Fc were diluted in phosphate-buffered saline (PBS) and used in the absence of adjuvant. Purified GP5 was emulsified with Freund's complete adjuvant for the primary immunization, and mixed with Freund's incomplete adjuvant for the boost injections. Mice were sacrificed 7 days after the last immunization, and serum samples were collected for further analyses. Serum from unvaccinated mice was collected and used as negative control.
Virus purification
PRRSV was propagated in MARC-145 cells, which were grown in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum at 37°C in 5% CO2. At 4 days postinfection of PRRSV, cells were frozen and thawed three times, and the culture supernatant was clarified by centrifugation at 17,132×g for 10 min. PRRSV particles were purified from the supernatant by ultracentrifugation at 134,320×g (Beckman, SW41Ti) for 2 h on a 20% sucrose cushion. The virus pellet was resuspended in PBS and stored in aliquots at −80°C.
Indirect enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay (ELISA) plates were coated with ∼5 μg/mL antigen diluted in 50 μL of 50 mM sodium carbonate buffer (pH 9.6) overnight at 4°C. The plates were washed three times with PBS with 0.05% Tween-20 (TPBS) and blocked with 5% nonfat milk in TBST buffer for 1 h at 37°C. After three washes, twofold serial diluted serum samples were added to incubate with the antigen for 1 h at 37°C. After washing five times, HRP-conjugated goat anti-mouse IgG (1:2,000 diluted in blocking buffer) was added to each well and incubated for 1 h at 37°C. After washing with TBST, 50 μL of TMB was added into each well and incubated in the dark for 10–30 min at 37°C. The reaction was stopped by adding 50 μL of 1M H2SO4, and the absorbance was read at 450 nm. The endpoint antibody titers were determined for each group and expressed as the reciprocal of the highest dilution of sera producing ratio values of 2.1.
Western blot
Proteins were separated by 12% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with 5% nonfat milk in TBST buffer at 4°C overnight, and incubated with the serum samples from the mice in TBST for 1 h at 37°C. Following three washes with TBST, the membrane was incubated with HRP-conjugated goat anti-mouse IgG (Beijing CoWin Biotech) and bound antibodies were detected using chemiluminescence luminol reagents (eECL Western Blot Kit).
Virus neutralization test
The serum samples collected from the mice were twofold serial diluted in DMEM after heat-inactivating for 30 min at 56°C, and mixed with PRRSV (200 TCID50) and incubated at 37°C for 1 h. The mixtures were then added onto the MARC-145 cell monolayer and incubated at 37°C in 5% CO2. The cells were examined daily for 8 days for the appearance of cytopathic effects (CPE). The neutralization antibody titer was determined for each group and expressed as the reciprocal of the highest serum dilution in which no obvious CPE was observed.
Statistical analysis
All data were expressed as means with standard deviation (SD). p-Values were calculated by a two-tailed unpaired t-test using the Prism 6 program (GraphPad Software). p-Values <0.05 were considered statistically significant.
Results
Expression and purification of recombinant proteins
The truncated GP5 gene in the prokaryotic expression vector encoded a polypeptide of 119 amino acids containing the GP5 ectodomain (S32 to K59), a GGGGS×3 linker, the GP5 endodomain (C131 to L200), and a 6-histidine tag on the C terminus. SDS-PAGE analysis showed that the protein purified from E. coli cells was about 13 kDa (Fig. 1B, lane 1), corresponded to its predicted molecular weight.

Expression and purification of GP5 proteins.
GP5-Fc and GP5N-Fc were produced using a baculovirus/insect cell expression system. With a C-terminal Fc tag, the unmodified proteins were predicted to have molecular weights of 41 kDa and 31 kDa respectively. When expressed in insect cells, several extra kilo Daltons of glycans could be added to the proteins at three potential glycosylation sites on the GP5 ectodomain and another one on the Fc tag. Western blot analysis detected unglycosylated, partially glycosylated intermediates, and fully glycosylated proteins in the cell extracts (Fig. 1A, lanes 2 and 4), while only glycosylated mature proteins were secreted into the supernatants (Fig. 1A, lanes 1 and 3). Purified GP5N-Fc had a molecular weight of about 40 kDa (Fig. 1B, lane 3), consistent with the predicted size for the fully glycosylated form of the protein. Purified GP5-Fc showed a predominant band at 45 kDa and a weak bigger band (Fig. 1B, lane 2), which may represent different glycosylated products of this protein.
Immunogenecity of recombinant proteins
To evaluate the immunogenicity of the GP5 proteins, BALB/c mice were respectively immunized with purified GP5 protein with adjuvant, GP5-Fc, or GP5N-Fc in the absence of adjuvant, and the antisera were collected 7 days after the last immunization. GP5-specific antibodies were examined and detected in all the three serum samples using the GP5 purified from E. coli cells as the antigen to coat the ELISA plate. The antibody titer of GP5 antiserum was more than 100 times higher than GP5-Fc and GP5N-Fc antisera (Fig. 2A), but it could be overestimated here, as antibodies to bacterial contaminant proteins could be elicited and then detected by the bacterium-derived antigen. Western blot confirmed that GP5 antiserum showed stronger reactivity to its immunogen than the other two antisera, although the differences seemed less distinct than in ELISA (Fig. 2B).
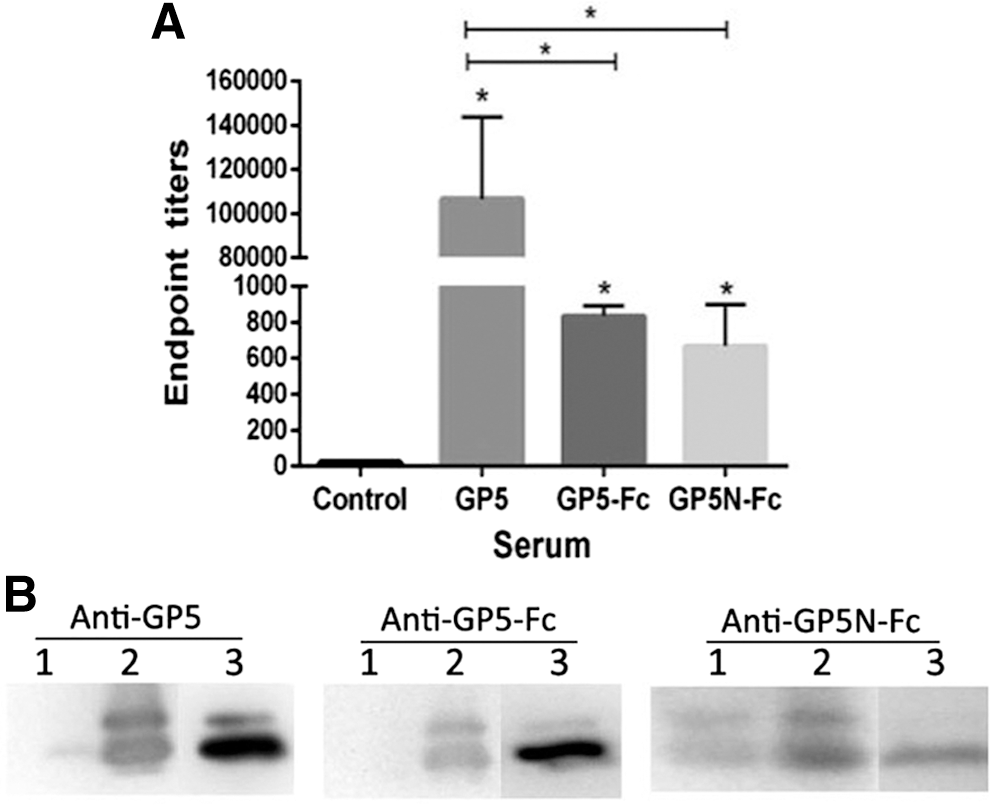
Analysis of serum antibody reactivity to bacterium-derived GP5.
Using purified GP5N-GST and GP5C-GST fusion proteins as antigens to assess the antisera further, bacterium-derived GP5 elicited strong immune responses to both the ectodomain and endodomain of the protein, which confirmed that the bacterium-derived GP5 was a good immunogen to elicit GP5-specific antibodies. GP5-Fc antiserum showed stronger reactivity to the ectodomain than the endodomain. GP5N-Fc only induced responses to the ectodomain as expected, but the titer of the antibody against the ectodomain was higher than GP5-Fc antiserum (Fig. 3), suggesting that the ectodomain was more immunogenic when it was directly fused with Fc tag in GP5N-Fc than in GP5-Fc.
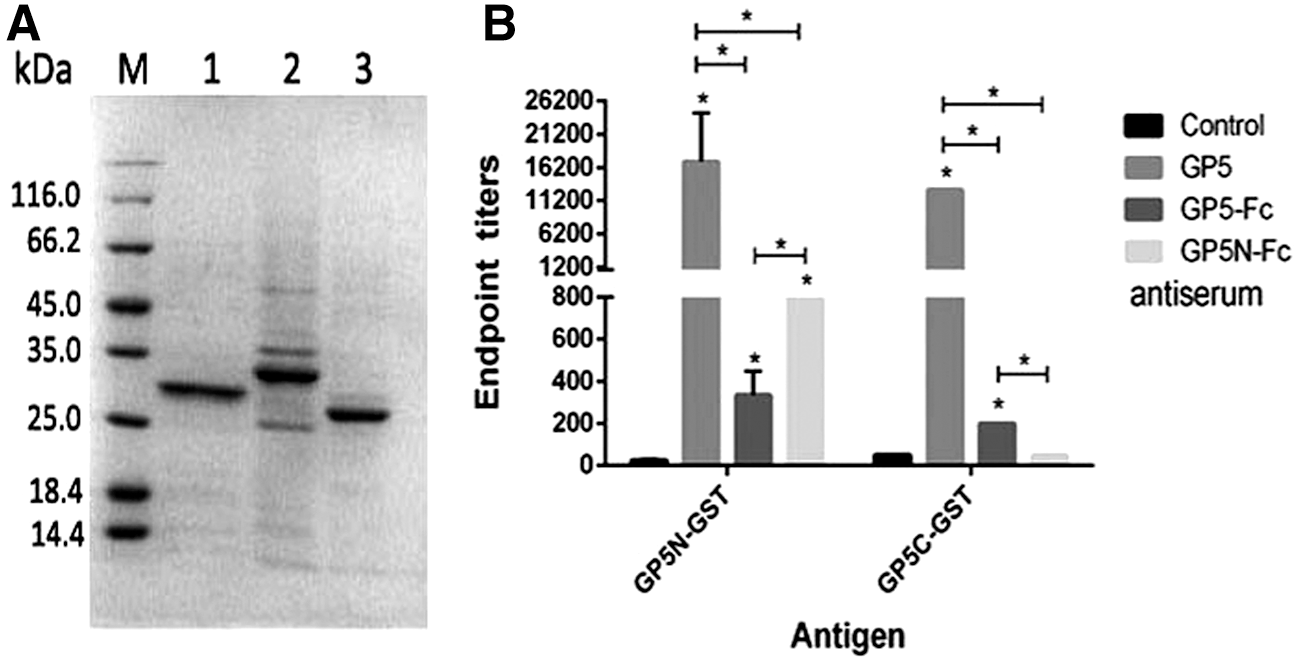
Analysis of serum antibody reactivity to the GST fused ectodomain and endodomain of GP5.
When purified PRRSV particles were used as the coating antigen to measure PRRSV-specific antibodies by ELISA, a high titer of antibodies was detected in every immunized group, and the GP5-Fc antiserum showed the strongest reactivity, although the differences between the immunized groups were not statistically significant (Fig. 4A). By Western blot, GP5 antiserum reacted best among the three groups to the denatured GP5 in purified PRRSV virions (Fig. 4B). These results suggested that bacterium-derived GP5 was probably more immunogenic than insect cell-derived GP5-Fc and GP5N-Fc, and it could induce more antibodies recognizing sequential epitopes on GP5, but GP5-Fc and GP5N-Fc were also good immunogens for inducing antibodies reactive to the native GP5 present on the surface of the virus particles.
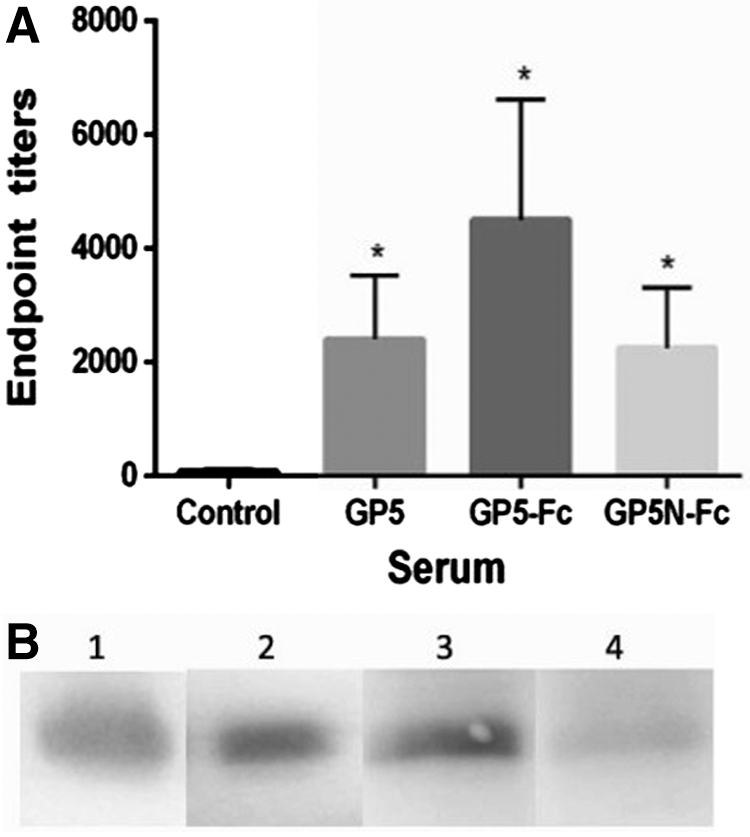
Analysis of serum antibody reactivity to purified PRRSV.
Virus neutralization assay
The neutralization titers of the serum samples were detected with virus neutralization assay in MARC-145 cells. As shown in Figure 5, bacterially expressed GP5 induced significant titer (p<0.05 vs. unimmunized control serum) of neutralizing antibodies that inhibited PRRSV infection in cells. This result is consistent with a previous report that the reengineered GP5 fusion protein with the deletion of the signal peptide and transmembrane regions could elicit neutralizing antibodies against PRRSV. Insect cell-derived GP5-Fc also elicited neutralizing antibodies, at a titer similar to the bacterium-derived GP5 protein, suggesting that it could be a potent immunogen for inducing protective immune responses against PRRSV infection. Although the neutralizing epitopes of GP5 have been mapped to the ectodomain and fusion of the ectodomain to Fc tag elicited predominant responses to the ectodomain, GP5N-Fc failed to induce significant neutralizing antibodies against PRRSV (p>0.05 vs. unimmunized control serum).

PRRSV neutralization assay. Marc-145 cells were infected with TA-12 strain of PRRSV, which was pretreated with the indicated antiserum. The neutralizing antibody titers were counted using the reciprocal of highest serum dilution in which no obvious CPE was observed in the well. All samples were analyzed in duplicate and the results were expressed as the average of the duplicates. *p<0.05 vs. unimmunized control serum. *p<0.05 between the marked immunized groups.
Discussion
Neutralization antibodies play an important role in the protection of pigs against PRRSV infection (15). As the most abundant envelope glycoprotein and a key inducer of neutralizing antibodies, PRRSV GP5 is considered a leading target for developing a subunit vaccine. However, it has been demonstrated that the neutralizing epitopes in the short ectodomain of the protein are poorly immunogenic due to the presence of a non-neutralizing decoy epitope (amino acids 27–31) (22) and shielding N-linked glycans nearby. Previous studies have shown that optimal modification of the protein, by addition of extra sequences between the neutralizing and non-neutralizing decoy epitopes, abrogation of the non-neutralizing decoy epitope by mutation and/or removal of some glycosylation sites (1,6,7,14,29), could enhance the immunogenicity of GP5.
Recently, a bacterium-derived GP5 with the deletion of its signal peptide and transmembrane regions was reported to be reactive with anti-PRRSV antibodies, and capable of stimulating the production of neutralizing antibodies in rabbit, which could inhibit the PRRSV infection in Marc-145 (26). In the present study, we engineered a similar GP5 protein, containing the ectodomain neutralizing epitope (S32 to K59) and the endodomain (C131 to L200) linked with a (GGGGS×3) linker, using a synthesized gene based on the GP5 sequences of a highly pathogenic PRRSV strain. Our results confirmed the previous report that the redesigned bacterium-derived GP5 was a good inducer of GP5 specific antibodies, and it could also induce a significant titer of PRRSV neutralizing antibodies in mice.
To improve the immune properties of the engineered GP5 protein further and focus the immune responses on the ectodomain, two more GP5 proteins fused with Fc tag (GP5-Fc and GP5N-Fc) were designed and produced using baculovirus/insect cell expression system in this study. Compared with the bacterium-derived GP5, the insect cell-derived glycoproteins induced significantly lower titers of GP5 specific antibodies (Fig. 2), suggesting that these glycoproteins were less immunogenic than the bacterium-derived GP5, even with the Fc tag to improve their immunogenicity. Nevertheless, both GP5-Fc and GP5N-Fc elicited strong serum responses to the native GP5 protein present on the surface of PRRSV virons (Fig. 4), and immunization of GP5-Fc resulted in slightly higher titer of neutralizing antibodies than the bacterium-derived GP5 (Fig. 5), although this result was not statistically significant. It has been demonstrated that the presence of neutralizing antibodies at a titer higher than 1:8 can protect pigs against PRRSV viremia (15). GP5-Fc induced neutralizing antibodies at a titer of 1:20 in mice in the absence of adjuvant (Fig. 5), indicating that this Fc tagged GP5 glycoprotein could be a potential candidate for the development of adjuvant-free subunit vaccines targeting the humoral immunity.
The neutralizing epitopes of PRRSV GP5 have been defined as linear antigenic peptides located in the ectodomain, but previous use of peptides resembling the GP5 neutralizing epitope failed to stimulate protective antibodies (16). When GP5 and M proteins were co-expressed as immunogen, the GP5-M protein complex produced by two different promoters was more immunogenic than the GP5-M fusion protein (38,39), and insertion of a GPGP linker between GP5 and M improved the immunogenicity of the fusion protein (4). In this study, fusing Fc to the ectodomain of GP5 (GP5N-Fc) elicited substantial immune responses to the ectodomain but failed to induce a significant titer of neutralizing antibodies, despite the fact that the neutralizing epitopes only locate in the ectodomain. In contrast, GP5-Fc induced a significant level of neutralizing antibodies. The presence of the endodomain in GP5-Fc may benefit the folding of the ectodomain to form a proper conformation to induce the production of PRRSV-specific neutralizing antibodies.
N-glycosylation is a common post-translational modification in eukaryotic cells. For vaccine immunogens, N-glycan modification usually plays an essential role in the maturation and correct folding of glycoproteins, but on the other hand, it may reduce the immunogenicity of the proteins. In the case of PRRSV GP5, heavy glycosylation in the ectodomain was considered as a way to shield the protein from recognition by the immune system, and deletion of some N-glycosylation sites could enhance the immunogenicity of GP5 (1,6,14). However, N44 glycosylation has been found to be essential for virus infectivity (1) and responsible for the rapid inducing neutralizing antibodies. In the present study, the glycosylated GP5-Fc was shown to be a potent inducer of neutralizing antibodies and serum responses to the native GP5 at least as good as the non-glycosylated GP5, although nonglycosylated GP5 was more immunogenic and elicited much higher level of GP5-specific antibodies. These results suggest further that proper folding of the protein via glycan modification may contribute to the effective presentation of the neutralizing epitopes to the immune system.
In summary, we show here that a reconstructed GP5 glycoprotein, containing the ectodomain and endodomain fused with an Fc tag, is an effective inducer of serum responses to the native GP5 on virus particles and neutralizing antibodies against PRRSV in mice. This designed immunogen may have the potential to be a candidate for the future development of new generation vaccine against PRRSV.
Footnotes
Acknowledgments
This project is supported by the Fundamental Research Funds for the Central Universities (QN2011065), and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry.
Author Disclosure Statement
No competing financial interests exist.