
Abstract
Cell mediated immune response plays a key role in combating viral infection and thus identification of new vaccine targets manifesting T cell mediated response may serve as an ideal approach for influenza vaccine. The present study involves the application of an immunoinformatics-based consensus approach for epitope prediction (three epitope prediction tools each for CD4+ and CD8+ T cell epitopes) and molecular docking to identify peptide sequences containing T cell epitopes using the conserved sequences from all the Matrix 1 protein sequences of H1N1 virus available until April 2015. Three peptides comprising CD4+ and CD8+ T cell epitopes were obtained, which were not exactly reported in earlier studies. Population coverage study of these multi-epitope peptides revealed that they are capable of inducing a potent immune response belonging to individuals from different populations and ethnicity distributed around the globe. Conservation study with other subtypes of influenza virus infecting humans (H2N2, H5N1, H7N9, and H3N2) revealed that these three peptides were conserved (>90%), with 100% identity in most of these strains. Hence, these peptides can impart immunity against H1N1 as well as other subtypes of influenza virus. A molecular docking study of the predicted peptides with class I and II human leukocyte antigen (HLA) molecules has shown that the majority of them have comparable binding energies to that of native peptides. Hence, these peptides from Matrix 1 protein of H1N1 appear to be promising candidates for universal vaccine design.
Introduction
T
As an internal protein, M1 appears to be less immunogenic, especially in the production of antibodies, compared with surface hemagglutinin (HA) and neuraminidase (NA) proteins. But in recent years, this protein has been targeted to develop a universal influenza vaccine (3,12,38). In a recent study conducted by Wang et al. (50), mice vaccinated with DNA plasmids and recombinant vaccinia virus expressing the M1 protein in combination with other conserved proteins (nucleoprotein and polymerase basic protein 1) were completely or partially protected after challenge with a lethal dose of influenza A virus (strain A/Puerto Rico/8/1934 H1N1). In another study, HA and M1 proteins (H5N1) expressed in Pichia pastoris were purified and used to immunize mice. A higher antibody mediated immune response was observed in mice against rHA and rM1 than mice immunized with either HA or M1 antigens (44).
The M1 protein is transcribed and further translated as one of the proteins coded by segment seven of the influenza A genome, along with other two splice variant: M2 ion channel and M42 (53). The M1 monomer (252 amino acid) is 60 Å long (40), carrying two globular regions—the N-terminal (1–164) and C-terminal (165–252)—linked by a protease-sensitive loop. Crystallographic studies have revealed that the M1 protein is composed of nine α-helices (H1–H9). These α-helices are organized into two four-helix bundles: N-terminal domain (H1–H4) and C terminal domain (H6–H9) connected via another helix linker (H5). The M1 protein lies beneath the viral envelope and plays diverse roles in the life cycle of the influenza A virus. It binds simultaneously to the viral envelope and RNA. It encapsulates the ribonucleoprotein (RNP) complex of influenza virus in the membrane envelope, thus stabilizing the membrane architecture. The M1 protein is also assumed to form cross-links with the cytoplasmic tail of HA and NA proteins, and commences viral budding from the host cell. During the virus entry, low pH induces a structural transition in the M1 protein inside the endosomal compartment, leading to the formation of unstable M1 monomers. This dislodging of the M1 protein shell expedites the release of vRNPs into the cytoplasm. Viral RNPs thus enter the nucleus and initiate the transcription and replication of the viral genome. The M1 protein in association with nuclear export protein (NEP) also facilitates the nucleocytoplasmic transport of newly formed vRNPs. Influenza virus exists in two different morphological forms: spherical (∼100 nm diameter) and filamentous (∼100 nm × 2–20 μm) virions (18). The filamentous trait of influenza virus has been traced to helix 6 domains of the M1 protein (8). The helix 6 domain (91–105 amino acids) of the M1 protein has been recognized as the multifunctional domain acting as a nuclear localization signal (NLS) for the translocation of the M1 protein in the nucleus and transcription inhibition motifs. Thus, the M1 protein plays multiple roles in influenza virus replication, from virus entry, uncoating, assembly, and budding of the virus particle.
Adaptive immune response plays a vital role in viral clearance, and thus highlights the importance of T cell epitope identification among various influenza virus proteins. Humoral immune response helps in influenza virus clearance mainly via complement dependent lytic antibodies (54). Cell mediated immune response targeted toward viral peptides, which are likely to be shared between different virus strains, may offer large dimensions to the immunity imparted against various strains and subtypes of influenza virus. Considering this fact, the current study is focused on identification of conserved immunogenic peptides comprising multiple T cell epitopes from the M1 protein of H1N1 influenza virus by applying a combination of various immunoinformatics tools that may be interesting candidates for a peptide-based universal influenza vaccine. Further, molecular docking was performed with the identified immunogenic peptide to calculate the binding affinity with different human leukocyte antigen class I and II molecules.
Materials and Methods
Sequence retrieval and conservation analysis
In the current study, full-length M1 protein sequences of H1N1, infecting humans all over the world, were retrieved until April 2015 from the Influenza Research Database (IRD; NIH/DHHS). MUSCLE and AVANA tools were used to obtain the conserved peptide stretches (≥90%) (17,27,28). Conservation refers to identical peptide sequences that are present in at least 90% of total M1 protein sequences, and these conserved peptide sequences were used for further epitope prediction. Full-length M1 protein sequences identified from human isolates of H5N1, H3N2, H7N9, and H2N2 were also retrieved from the IRD for conservancy analysis.
Identification of T cell epitopes
Performance of the consensus prediction approach of epitope prediction was found to be superior to the single predictive strategy (29). In our earlier report, the consensus approach has been used for epitope prediction of H1N1 HA and NA proteins (28). The current study also employed different prediction tools: NetCTL 1.2, SYFPEITHI, and BIMAS (26,34,39) for class I binding (CD8+ T cell) epitopes and Immune Epitope Database (IEDB)–based SMM align, ProPred and NetMHC II for class II binding (CD4+ T cell) epitopes (32,33,43). Threshold scores taken for the class I epitope prediction tool, SYPFEITHI, Net-CTL, and BIMAS were 20, 0.75, and 20, whereas for class II HLA prediction tools, SMM align, and NetMHC II, it was IC50 ≤ 500 nM. All the HLA alleles/supertypes provided in each prediction tool were considered for epitope prediction (Table 1). NetCTL predicts epitopes for HLA supertypes in place of alleles. Each supertype represents a set of HLA alleles that share largely overlapping peptide-binding specificity. Class I HLA has been classified into 12 different supertypes. As per the criteria defined in a previous report (27), peptides were defined as epitopes, and further epitopes showing identity to the human proteome based on pBLAST (2) were eliminated from further studies. The final conserved epitopes showing an overlap were merged into a single peptide.
NetCTL predicts epitopes for HLA supertypes in place of alleles. Each HLA supertype represents a set of HLA alleles that share largely overlapping peptide binding specificity.
Number signifies the number of HLA alleles/supertypes.
HLA, human leukocyte antigen.
HLA distribution analysis
An epitope can elicit an immune response in individuals expressing the HLA capable of binding that epitope. HLA polymorphism exists among different geographical areas and ethnicities. Thus, selecting epitopes with binding affinity with multiple HLA alleles increases the coverage of the population targeted by peptide-based vaccines. The IEDB population coverage analysis tool is based on an allele frequency database that provides allele frequencies for 115 countries and 20 different ethnicities grouped into 16 different geographical areas (7). Analysis of the peptides was carried out for the populations distributed in 16 geographical zones, as well as 20 ethnic groups all over the world.
Molecular docking of peptides comprising epitopes
High-resolution crystallographic structures of 10 class I HLA and five class II HLA molecules were fetched from the protein data bank. These HLA molecules represented various supertypes of class I and II HLA (13,42). The naturally bound peptides (native peptides) of the HLA molecules were cut apart using the Discovery studio visualizer (v4.1) and docked again using the AutoDock Vina tool as the test set (49). The grid for each of the HLA molecules was defined based on native peptides. The structure of the peptides comprising epitopes was generated using the PEP-FOLD server (48). These generated structures were used for docking with class I and II HLA molecules.
Results
Conserved peptides of the M1 protein in H1N1 virus
To cover all the genetic variants of H1N1 isolates around the world, 458 sequences of the M1 protein (human host) were obtained after removing the duplicate ones. Frequent mutations in influenza virus render previously identified epitopes non-immunogenic. Thus, peptides of the M1 protein conserved among all the strains of H1N1 can prove to be more effective candidates of immunogenicity. Ten conserved peptide sequences of the M1 protein (M1.1–M1.10) were identified using AVANA, and ranged in size from 12 to 49 amino acids (Table 2). The first seven (M1.1–M1.7) conserved peptides are confined to the membrane-binding domain of the M1, whereas the last three (M1.8–M1.10) conserved peptides are located in the RNP-binding domain.
Identification of peptides comprising CD4+ and CD8+ T cell epitopes
Cell mediated immunity plays an important role in controlling influenza infection. Hence, the T cell epitope prediction was carried out based on different immunoinformatics tools for CD8+ T cell epitopes (NetCTL, BIMAS, and Syfpeithi) and CD4+ T cell epitopes prediction (NetMHC II, ProPred, and IEDB-based SMM align). After comparing the predicted epitopes with the respective HLA restriction, a set of epitopes was obtained that was predicted by all the tools separately for class I and II HLA. BLAST analysis revealed no specific homology of the human proteome with any of the predicted CD4+ T cell epitopes, but among CD8+ T cell epitopes of the M1 protein, one epitope was found to be homologous as per the set criterion, so it was eliminated from further studies (data not shown). Finally, 10 CD8+ T cell epitopes (class I HLA binding epitopes) and nine CD4+ T cell epitopes (HLA class II binding epitopes) of the M1 protein were considered for further analysis (Table 3). In Net MHCII 2.2 and IEDB-based SMM align, the binding affinity of the CD4+ T epitope to HLA is given as IC50. For each predicted epitope, the IC50 value varies with various combinations of HLA alleles and tools used. The range of IC50 values for predicted CD4+ T cell epitopes is given in Table 3 to show their binding affinity with HLA. Epitopes MVLASTTAK and IRHENRMVL of the M1.8 conserved sequence were predicted to bind to both class I and II HLA molecules. Epitopes having overlapping amino acid sequences in the respective conserved regions were merged to generate a longer peptide having multiple epitopes. In this way, six peptides of CD8+ T cell epitopes (length 9–15 amino acids) and three peptides containing CD4+ T epitopes (length 12–17 amino acids) were obtained (Table 4). The overlapping epitopes in these peptides belong to either the same or different alleles/supertypes (Table 4). The epitopes belonging to same versus different alleles/supertypes are represented as common versus unique alleles/supertypes, respectively (Table 4). Further, these putative immunogenic peptides were merged to generate a single peptide containing multiple CD4+ and CD8+ T cell epitopes, confined to a particular conserved region. Finally, three peptides (one peptide in the membrane binding domain and two peptides in the RNP binding domain) were obtained that contained both CD4+ and CD8+ T cell epitopes (Table 5).
Range of IC50 value for predicted epitopes with different HLA alleles using NetMHCII 2.2 and IEDB-SMM align.

Conservation of peptides containing multiple epitopes among other subtypes of influenza A virus
Three final selected peptides containing multiple CD4+ and CD8+ T cell epitopes were searched for 100% identity as a measure of conservation in seasonal H1N1, pandemic H1N1, H5N1, H3N2, H2N2, and H7N9 M1 protein sequences. The first peptide (ILGFVFTLTVPSERGLQRRRF) is conserved (≥90.9%) among all the subtypes of the influenza A virus (Table 6). The second (LIRHENRMVLASTTAKA) and third (LQAYQKRMGVQMQR) peptides were also found be conserved (≥90%) in most of these subtypes (Table 6). High conservation was not observed for the second and third peptides in H3N2. Hence, identity consideration was relaxed to ≥90%, and it was then found be conserved (≥97%). In H7N9, the third peptide (LQAYQKRMGVQMQR) was replaced by LQAYQNRMGVQLQR with mutation at the sixth and twelfth position in all the sequences. Additional data containing multiple sequence alignment showing sequence identity of the three final selected peptides in seasonal H1N1, pandemic H1N1, H5N1, H3N2, H2N2, and H7N9 are given in Supplementary Tables S1–S6 (Supplementary Data are available online at
Conservations were recorded at 100% sequence identity.
Number of full-length, nonredundant sequences of the M1 protein available in the database until April 2015.
Conservation at identity ≥90%.
Population coverage of HLA predicted to bind M1 peptides
HLA polymorphism among populations from different geographical areas and ethnicities results in a variation of the peptide's immunogenic response. Hence, population coverage analysis becomes important. Population coverage analysis of the three M1 peptides was carried out for populations distributed in 16 geographical zones and 20 ethnicities (Fig. 1). The average population coverage was observed to be 98.13% jointly for class I and II HLA binding peptides of M1. Among the different geographical zones, 100% of populations of South Asian, European, North American, and East, West, and Central African populations are expected to respond to these predicted epitopes. Further, HLA distribution analysis was also carried out among the different ethnicities distributed worldwide, which resulted in an average of 98.61% coverage of HLA (Fig. 1B). Among various races, Asians, blacks, Caucasians, Melanesians, and Siberians showed 100% coverage.
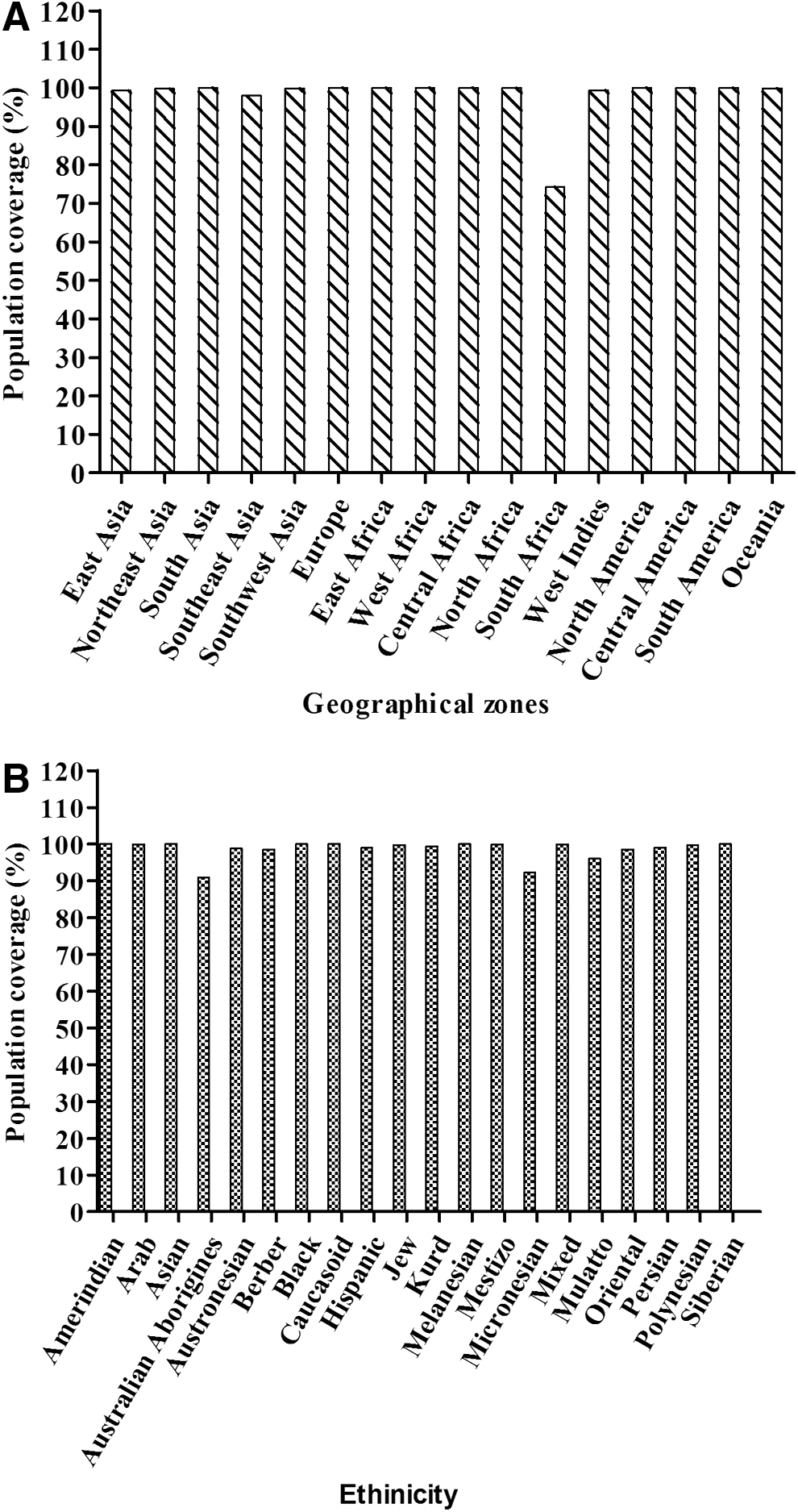
Population coverage analysis of predicted Matrix 1 (M1) peptides showing the expected response. (
Peptide docking with class I and II HLA molecules
The first and most important step toward the generation of an efficient adaptive immune response is the binding of the immunogenic peptide to the peptide-binding groove of the HLA molecule inside the host cell. The AutoDock Vina tool was employed to calculate the binding affinity of the peptide-containing epitope with class I and II HLA molecules. The crystallographic structures of peptide bound ten HLA class I (resolution 1.3–2 Å) and five HLA class II molecules (resolution 1.7–2.75 Å) were taken from protein data bank (PDB id mentioned in Tables 7 and 8) for docking. Each of the HLA molecules represents different HLA supertype (13, 42). Native peptides were separated from the respective HLA molecule and docked to obtain the binding energy that was used as a test set for comparison. Since class I HLA is known to bind short peptides (length 9 amino acids), the predicted five class I HLA peptides (part of three final peptides containing multiple epitopes) were used for docking with the class I HLA molecules. Considering that class II HLA can accommodate bigger peptides, three peptides containing multiple epitopes (part of three final peptides) were docked with the class II HLA molecules. The PEP-FOLD generated structures of these peptides containing epitopes were docked with different HLA I and II molecules. It was observed that most of the peptides showed comparable binding energy to that of the native peptides with respective HLA molecules (Tables 7 and 8). However, some of the peptides were found to bind outside the peptide-binding groove of HLA, which were designated as nonbinders (NBs; Tables 7 and 8). The binding energy of these peptides was spotted within the range of native peptides for both class I and II HLA molecules (Fig. 2A and B). Dunnett's multiple comparison test was carried out to compare the binding energy between these peptides and native peptides as the control peptide. Binding energies of three out of five peptides for class I HLA and all three peptides for class II HLA were not found to be significantly different from respective control peptides. Binding energies of the MVLASTTAK and KRMGVQMQR peptides appear to be significantly less compared with the control peptide. Furthermore, analysis of these results showed that the MVLASTTAK and KRMGVQMQR peptides were not binding in the peptide-binding groove of five and two HLA class I molecules. The peptide containing the MVLASTTAK epitope also did not fit in the binding groove of three class II HLA molecules. Hence, this peptide may be not a good binder for either class I or II HLA.
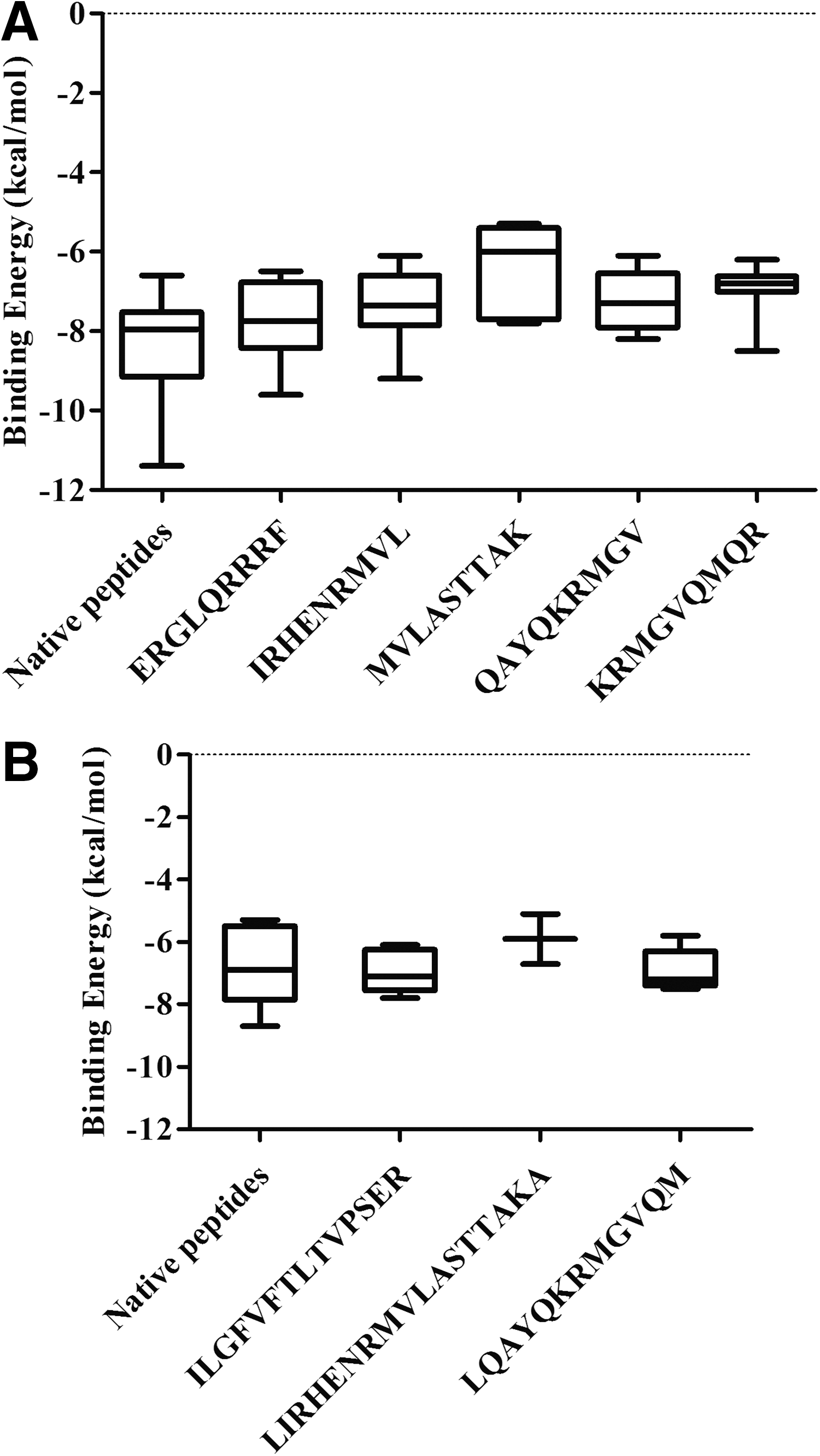
Binding energy of the predicted peptides and native peptides after docking with (
Discussion
The availability of a substantial amount of data of relevance to immunology research, particularly clinical and epidemiologic data, has pioneered the contemporary domain of immunoinformatics or computational immunology. Immunoinformatics-guided epitope prediction (T and B cell) is speculated to improve the selection of targets for vaccine design by reducing the time and cost of laboratory identification of epitopes. Studies undertaken against Mycobacterium tuberculosis (24) and Leishmania amazonensis (36) have demonstrated the efficiency of computationally predicted epitopes to evoke an immune response both in vitro and in vivo. Furthermore, studies conducted against influenza virus proteins by Duvvuri et al. (15) and Sun et al. (46) established the success of computational approaches in predicting epitopes. In these studies, the predicted influenza epitopes prove to be immunogenic in the case of healthy human peripheral blood mononuclear cells and murine models.
Genome- or proteome-wide in silico screening of severe and ever-changing entities such as the influenza virus for putative immunogenic peptides can accelerate the global pursuit of universal influenza vaccine development. Incessant mutations (antigenic shift and drift) in influenza forfend the immunogenic potency of predefined epitopes against all the variants of a particular subtype of influenza virus, as well as other subtypes in case of cross-reactive epitopes. Hence, targeting conserved peptide sequences to identify the epitopes appears to be an elite approach for vaccine design. The current study involved 458 unique protein sequences of H1N1 M1 (including seasonal as well pandemic strains of H1N1) deposited worldwide until April 2015, which is the largest as far as it is possible to tell from the literature for the identification of T cell epitopes from the M1 protein (H1N1). This is in contrast to earlier studies undertaken to predict T cell epitopes of the M1 protein in H7N9 and H3N2 (14,16,21), which considered reference strains for epitope prediction.
The current study was undertaken using a consensus approach of epitope prediction, which employs different prediction programs in order to define the epitopes (27). Use of a single computational algorithm for T cell epitope prediction may lead to a substantial number of false positives and false negatives. Taking this into consideration, the consensus approach (i.e., combining and comparing the different prediction algorithms to identify epitopes) has been applied in various studies, and its immunogenic potential has been verified in vitro and in vivo (11,41). The immunogenicity of peptides depends on multiple factors mediating peptide immune response, such as HLA-peptide binding, TAP transport efficiency, C-terminal cleavage, and binding stability of the peptide–HLA complex. Considering this, multiple prediction tools for CD8+ T cell epitopes (NetCTL, BIMAS, and SYFPEITHI) and CD4+ T cell epitopes (ProPred, SMM align, and NetMHC II v2.2) were employed, which are based on different immunogenic factors and algorithms. NetCTL v1.2 is an Artificial Neural Network (ANN)–based tool that predicts CD8+ T cell epitopes taking into account TAP transport efficiency and C terminal cleavage, while BIMAS identifies epitopes based on the measurement of the dissociation half-life of (t1/2) of β2 microglobulin. The SYFPEITHI is a continuously updated database of MHC ligands and peptide motifs of human and other species, which predicts epitopes based on reported motifs. ProPred employs a literature-based amino acid/position coefficient table, and presents the HLA-DR binding propensity of given protein sequence (43). SMM align and NetMHC II v2.2 employs a position-specific weight matrix and ANN, respectively, for binding affinity to different HLA-DR, and their binding affinity is given as IC50 values. These T cell epitope prediction tools have been extensively used and their efficacy has been verified experimentally by various studies (9,11,20,41). In addition to the multi-immunogenic factor and different prediction algorithms, the consensus approach also offers advantages with regard to predicting large numbers of different HLA alleles.
Earlier studies have reported partially identical stretches of peptides between human and influenza proteins (21). Any kind of molecular mimicry between the predicted epitopes and host protein (human) may lead to cross-reactivity and, further, autoimmune response. One predicted epitope of the M1 protein, GILGFVFTL, was found to be homologous to human tetraspanin-33 during BLAST analysis, which is accordance with an earlier report (21). It is considered to be immunodominant in HLA-A2 individuals, and it also overlaps with NLS (47). Although, it is one of the most extensively studied epitopes of the M1 protein, in this study it was still eliminated from further consideration to avoid any chances of molecular mimicry.
Vaccines induce virus-specific antibodies that bind specifically to the pathogen and mediate effector functions, thus limiting the infection. Normally, the surface proteins of the virus are the prime targets of antibodies secreted by B cells. However, frequent mutations in influenza A virus strains render these pre-existing antibodies ineffective over time. Prototypically, the virus has been known to be eliminated by T cell mediated immune response. CD8+ T cells employ perforin, Fas/Fas-Ligand, and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) mediated mechanisms to kill virus-infected cells and thus clear viral infection (22). CD4+ T cells, however, contribute toward antiviral immune response primarily by assisting CD8+ T cells and B cells by releasing antiviral cytokines (1). Thus, a T cell epitope-based vaccination approach appears to activate both humoral and cell mediated arms of adaptive immunity to eliminate viral infection jointly.
Expanding knowledge about antigen presentation and recognition has facilitated the advancement in vaccine development, especially peptide-based vaccines. Peptide-based vaccines are favored because they lack the risk of reversion to virulent and genetic integration or recombination. Additionally, they are relatively easy to produce and can be modified to enhance their immunogenicity, stability, and solubility (37). Furthermore, T cell epitopes (peptides) are expressed on the surface of antigens, presenting as a peptide–HLA complex. Because of the high polymorphism in HLA among different populations and ethnic groups, a variable immunogenic response of some epitopes is expected. Thus, peptides capable of multiple HLA restriction or peptides bearing multiple epitopes are preferred as vaccine targets for extensive and impartial population coverage required for a vaccine. Peptides containing both CD4+ and CD8+ T cell epitopes may improve the immunogenicity and clinical outcomes of peptide-based vaccines (31). In silico epitopes prediction tools enable multi-epitope peptides to be selected in a prompt and cost-effective way, with vast coverage among the world's population. With this in mind, the current study reported three M1 peptides that were generated after merging the CD4+ and CD8+ T cell epitopes predicted by the immunoinformatics approach.
Certain groups of individuals are highly vulnerable to influenza infection and related complications, including children (<5 years), the elderly (>65 years), pregnant women, and people with special medical conditions such as asthma, AIDS, cancer, chronic lung disease, and so on. Furthermore, Blumenshine et al. (7) suggested that various factors determine the severity of pandemic influenza, including differential exposure to influenza due to income or ethnicity, unequal levels of susceptibility, differential access to prophylaxis, and treatment after disease develops. A study in the United States reported higher rate of hospitalization among Asian/Pacific Islanders, non-Hispanic blacks, and Hispanics compared with non-Hispanic whites during the 2009 pandemic (30). Another study showed a relatively high rate of pandemic (H1N1) fatality in Southeast Asians and Americans compared with Europeans and Africans (10). Globally, as reported by the World Health Organization (52), influenza activity has been higher in the southern hemisphere compared with the northern hemisphere. Moreover, the circulating strains of influenza keep mutating, thereby changing the susceptibility of a specific ethnic group toward influenza. Thus, there is an alarming need to view influenza from a global prospective. Interestingly, the population coverage analysis in the present study unveiled that predicted peptides showed an average population coverage of >98%, combining class I and II HLA epitopes among various geographical areas as well as ethnicities. Hence, these peptide sequences not only contain multiple T cell epitopes but also have the potential to induce a potent immunogenic response among individuals belonging to different populations around the world.
It was interesting to find a vaccine candidate that may be effective not only in one type of influenza virus but also in other subtypes. Hence, these identified peptides were studied for their conservation in other subtypes of influenza virus infecting humans (H2N2, H5N1, H7N9, and H3N2). This conservation study revealed that these three peptides were found to be >90% conserved, with 100% identity in most of the strains. These data suggest that these peptides can impart immunity against H1N1 as well as other subtypes of influenza virus (heterosubtypic immunity).
Molecular docking is employed not only for computer-aided drug designing, but also for peptide–HLA interactions. The docking approach was found to be reliable and accurate for evaluating peptide binding to HLAs (5,35). Peptides binding to class I HLA are normally nonamer, while for class II HLA they vary in length from 12 to 25 amino acids. Hence, peptides containing nonamer epitopes were docked with class I HLA, whereas longer sequences containing multiple epitopes were considered for docking with class II HLA. These peptide sequences were found to have comparable binding energy with that of natural bound peptides (test peptides) for both class I and II HLA, except that two class I HLA restricted MVLASTTAK and KRMGVQMQR. This could be because these epitopes failed to bind HLA molecules in the designated space, and thus were assumed to be NBs. Hence, the peptide containing this epitope may not have good affinity with either of the HLA molecules. This observation can be explained by the fact that the epitopes were not predicted to bind to all HLA molecules. However, most the peptides showed a good binding affinity with class I and II HLA molecules. A similar study was conducted, taking into account class I HLA alleles, in which nucleoprotein epitopes were identified and docked with four HLA-B molecules (23). In contrast, the present study included docking studies for both class I (10 molecules) and class II (five molecules) HLA molecules.
Databases such as the IEDB and the IRD provide complete information regarding experimentally determined epitopes via means of submission as well as available literature. Comparing conserved predicted CD4+ and CD8+ T cell epitopes with the experimentally determined epitopes from the data sets (IEDB) enabled the current study to assess the novelty of identified multi-epitope peptides. It provides an account of experimental data pertaining to antibody and T cell epitopes studied in humans, nonhuman primates, and other species. As of March 7, 2015, the IEDB reported 167 T cell epitopes and four linear B cell epitopes of the M1 protein in the human host. Previous findings suggest that the three peptides identified in the present study have not been reported, although some substrings of peptide fragments have been reported to induce a CD4+ and CD8+ T cell response (4,19,56). The literature reports that the substrings of these peptides bind specifically to only one or two HLA alleles, whereas the current study reports that these peptides are capable of binding to a large number of class I and II HLA alleles. Thus, this can be considered for experimental validation in vitro.
CD4+ T cells are considered to be an important factor in B cell activation and further antibody production. Hence, these peptides may also evoke a B cell response against the influenza virus. This is in accordance with an earlier study in which one of our identified peptides (LQAYQKRMGVQMQR) forms part of the B cell epitope, reported to induce IgG production in patients who have just recovered from H5N1 infection (25).
Internal proteins are more conserved than surface proteins. Hence, internal proteins such as the M1 protein and nucleoprotein are now being considered as an excellent target for a universal influenza vaccine. The M1 protein in various forms is being considered as a candidate for the generation of an adaptive T cell mediated immune response against influenza. In order to improve the immunogenic potential of an M1-based vaccine, several strategies have been suggested, including the administration of high-dose formulations of the vaccine, combination of modified vectored vaccine backbones, and adjuvants (3). In 2010, a study reported that the M1 protein expressed in Escherichia coli when used to immunize BALB/c mice by intranasal drip using chitosan adjuvant could protect 70% and 30% of the mice challenged with PR8 (H1N1) virus and A/Chicken/Henan/12/2004 (H5N1) virus, respectively (45). A novel influenza virus MVA-NP+M1 (modified vaccinia virus Ankara expressing conserved influenza internal nucleoprotein and M1 protein) targeting the M1 protein is under clinical trial (3). Although 167 linear M1 T cell epitopes have been reported in the human host, there is still a need for some probable peptide candidates in order to design a universal vaccine. The present study has identified three peptide sequences that contain these reported T cell epitopes based on immunoinformatics and molecular docking. These approaches serve as an initial screening and filtering step toward shortlisting potential immunogenic epitopes to synthesize for experimental validation, thus saving time, effort, and resources.
Footnotes
Acknowledgment
We gratefully acknowledge financial support from the DST, New Delhi, India (SERB/F/5066/2012-13).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.