
Abstract
Changes in interferon (IFN)-stimulated gene (ISG) expression in cells infected with measles virus (MeV), four wild strains (belonging to different genotypes), and the laboratory strain Edmonston were examined. ISGs [MxA, 2′-5′-oligoadenylate synthetase, and interferon regulatory factor-1] were upregulated in an MeV-infection-induced manner and in an IFN-induced manner. In MeV-infected SiHa cell lines, the MeV infection-induced expression levels were in the order of A>H1>D8>D5>D3. On the other hand, all infected cell lines abolished type I and III IFN-induced ISG expression. However, partial type II IFN-mediated induction was observed in the MeV-infected cells. The wild strain of genotype D3 was the most potent inhibitor of MeV infection-induced and IFN-induced ISG expression and generated the highest titer of infectious viral particles. Edmonston triggered the highest levels of MeV infection-induced ISG expression in SiHa cells and produced the lowest titer of infectious particles. Expression of the viral C protein was associated with suppression of MeV infection-induced and type II IFN-induced ISG expression.
Introduction
M
Measles virus (MeV) belongs to the genus Morbillivirus within the family Paramyxoviridae (8). There are two types of MeV strains; wild and laboratory. Wild strains infect human cells by signaling lymphocyte activation molecule (SLAM), which is expressed by activated B and T lymphocytes and dendritic cells, and by nectin 4, which is expressed by several epithelial cells (5,17). Laboratory strains, which are Vero cell-adapted viruses and belong to genotype A, infect through CD46, which is ubiquitously expressed by almost all eukaryotic cells, as an alternative receptor. Live attenuated vaccines are derivatives of genotype A.
We previously examined the mechanisms by which MeV suppresses interferon (IFN)-stimulated gene (ISG) expression as a counteracting strategy of host antiviral immune response (31 –33). We revealed that both a wild strain (AK1) and laboratory strains suppressed type I IFN signaling (33). However, MeV infection constitutively transduces IFN-γ-like signals through phosphorylation of signal transducer and activation of transcription 1 (STAT1), and the activated STAT1 induces expression of IFN regulatory factor-1 (IRF-1), thereby suppressing the growth of infected cells (31,32). MeV infection-induced IFN-γ-like signaling varies according to the viral strains.
The MeV C protein inhibits the IFN-γ-like signaling. Cells infected with the wild strain, AK1, express higher levels of C protein, leading to strong cell growth and the production of viral particles. We believe that all wild strains share this potent IFN-counteracting activity; however, we examined only strains harboring genotype D3 (31 –33). In this study, we examined how other genotypes of MeV wild strains [D5, D8, and H1] affect IFN-related antiviral system.
Materials and Methods
Cells and viruses
SiHa (a human cervical squamous carcinoma cell line) and Vero cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured in RPMI-1640 containing 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. Vero cell stably expressing human SLAM (Vero/hSLAM) was kindly provided by Dr. Yusuke Yanagi (Kyushu University, Fukuoka, Japan) and cultured in RPMI-1640 containing 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.4 mg/mL G418.
MeV strains Edmonston and AK1 have been described previously (31). Other wild strains, MVs/Hokkaido. JPN/52.07/11 [D5], MVi/Hokkaido. JPN/25.11 [D8], and MVs/Sapporo. JPN/19.10/1 [H1], were isolated from measles patients in Hokkaido prefecture, Japan, as described previously (12,14,15). Recombinant MeV strain IC323-EGFP, which contains the complete genome of wild strain IC-B [genotype D3] and the gene for green fluorescence protein, was donated by Dr. Yusuke Yanagi. Cells persistently infected with MeV were established according to the previous articles (7,33). Briefly, SiHa cells were infected with MeV at a multiplicity of infection (MOI) of 0.02 for 1 h at 37°C. The cells were then passaged every 3 or 4 days for 50 days. Cells infected with Edmonston were not passaged for 14 days postinfection due to the reduced number of live cells by cytopathic effect (CPE).
RNA isolation
Total cellular RNA and viral RNA in the culture supernatant were isolated using an RNeasy Mini Kit (QIAGEN) and a QIAamp Viral RNA Mini Kit (QIAGEN), respectively, according to the manufacturer's instructions.
Genotype of MeV
The MeV genotype was determined from a partial nucleotide sequence from the nucleoprotein (N) gene, which was obtained by direct sequencing of nested polymerase chain reaction (PCR) products (13). The primer sets used to detect the N gene were pMvGTf1m (5′-CGRTCTTACTTYGATCCRGC-3′) and pMvGTr1 (5′-TTATAACAATGATGGAGG-3′). Reverse transcription-PCR (RT-PCR) was carried out using a OneStep RT-PCR Kit (QIAGEN, Hilden, Germany). The RT-PCR products of the MeV N gene were amplified by nested PCR with HotStarTaq Master Mix (QIAGEN) and the following primer set: pMvGTf2m (5′AGAYTAGGRCARGAGATGGT-3′) and pMvGTr2 (5′-GAGGGTAGGCGGATGTTGTT-3′).
Nested PCR products were purified using a Wizard® SV Gel and PCR Clean-Up System (Promega, Madison, WI) and sequenced in an Applied Biosystems 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA) using a BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems). Nucleotide sequences from the partial MeV N gene (450 nucleotides) were aligned using the CLUSTAL W program on the DNA database of Japan (DDBJ; National Institute of Genetics, Mishima, Japan). A phylogenetic tree was constructed by the neighbor-joining method using MEGA6 software (28) with 1,000 bootstrap replicates.
Stimulation with IFN
Recombinant IFN-α2 was purchased from Serotec (Oxford, United Kingdom). Recombinant IFN-γ and recombinant IFN-λ1 were purchased from R&D Systems (Minneapolis, MN). SiHa cell and MeV-infected SiHa cells were inoculated in six-well plates, and incubated until 100% confluent. The cells were then stimulated with 103 IU/mL IFN-α2, 103 U/mL IFN-γ, or 10 ng/mL IFN-λ1, for 4 or 8 h. Nonstimulated cells were used as a control. After IFN treatment, the cells were washed with Dulbecco's phosphate-buffered saline (PBS) and stored at −80°C until total RNA purification.
Regents and antibodies
Ruxolitinib, a pan janus kinase (Jak) inhibitor that preferentially inhibits Jak1 and Jak2 (11,29), was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Ruxolitinib was routinely used at a concentration of 1 μM. Rabbit polyclonal antibodies against MeV C and V proteins were generated as described by Takeuchi et al. (27). The antibody against C protein reacts with wild strain one and laboratory strain one at a similar extent as previously described (31). Mouse monoclonal antibodies against MeV P, M and N, MxA, and β-actin and rabbit antibodies against STAT1 and Tyr-phosphorylated STAT1 were described previously (9,31,33). A mouse monoclonal antibody against GTPase-activating protein-binding protein 1 (G3BP1) was purchased from Abcam (Cambridge, United Kingdom).
Measurement of IFN-α, β, λ, and γ concentrations by ELISA
IFN-α was measured by using a Human Interferon Alpha All Subtype ELISA Kit (PBL, Piscataway, NJ). IFN-β was measured by using the Human Interferon-β ELISA kit (Kamakura Techno-Science, Kamakura, Japan). IFN-λ1, λ2, λ3, and γ were measured using the respective DouSet ELISA Development Kits (R&D systems).
Real-time PCR
cDNA was prepared from RNA using the Super Script®IIIFirst-Strand Synthesis System (Life Technologies, Grand Island, NY), according to the manufacturer's instructions.
cDNA was used as a template for real-time PCR. PCR was performed using a TaqMan® Gene Expression Master Mix (Life Technologies) in a LightCycler® 480 (Roche Diagnostic, Basel, Switzerland). Probes and primer sets targeting MxA (Hs00895608), 2′-5′-oligoadenylate synthetase (2′-5′-OAS) (Hs00973637), β-actin (Hs99999903), and IRF-1 (Hs00971960) (Life Technologies) were used. mRNA levels were normalized against β-actin. The expression levels of the target genes were calculated using the ΔΔCt method, according to the instructions supplied with the LightCycler 480 software (Roche).
Detection of defective interfering particle genes
Viral RNAs were purified from the MeV-infected SiHa cells and their culture supernatants, and RT-PCR was performed using a OneStep RT-PCR Kit.
Two sets of defective interfering (DI)-specific primers were used: the first set (4) composed A1, 5′-TCTGGTGTAAGTCTAGTATCAGA; A2, 5′-AAAGCTGGGAATAGAAACTTCG (targeting DI); and A2 and B1, 5′-ATGACAGATCTCAAGGCTAAC (targeting the full-length MeV genome): the second set (25,30) comprised JM396, 5′-TATAAGCTTACCAGACAAAGCTGGGAATAGAAACTTCG-3′; JM403, 5′-CGAAGATATTCTGGTGTAAGTCTAGTA-3′ (targeting DI); and JM396 and JM402, 5′-TTTATCCAGAATCTCAARTCCGG-3′ (targeting full-length MeV genome). The primer set targeting glyceraldehyde-3-phosphodehydrogenase (GAPDH) mRNA, which was used as a control for cellular RNA, has been described previously (33). The PCR products were analyzed by agarose gel electrophoresis.
Western blot analysis
Cells were scraped from the dish and collected in microcentrifuge tubes. Cells were lysed in a buffer comprising 50 mM HEPES-NaOH (pH 7.4), 1% Triton-X100, 0.5% sodium deoxycholate, 5% glycerol, 2 mM EDTA, 150 mM NaCl, and a protease inhibitor cocktail (Thermo Scientific, Pittsburgh, PA). Protein concentrations were determined using a BCA protein assay reagent kit (Thermo Scientific). Five to 30 μg of total protein/lane were separated by electrophoresis on 7.5%, 12.5%, or 15% sodium dodecyl sulfate–polyacrylamide gels (Wako Pure Chemical, Tokyo, Japan), followed by transfer to a polyvinyl difluoride membrane (Immobilon; Millipore, Bedford, United Kingdom) using a wet transfer equipment.
The membrane was saturated with a blocking buffer (Tris-buffered saline containing 0.1% Tween 20 and 1% skim milk or PBS containing 0.05% Tween 20 and 5% bovine serum albumin) for 1 h at room temperature and then incubated with anti-MeV V, P, N, M, or C protein antibody, or anti-β-actin antibody at room temperature or 4°C overnight. The membrane was then incubated with alkaline phosphatase conjugated anti-mouse and anti-rabbit IgG antibodies (Biosource International, Camarillo, CA) at room temperature for 1 h. Specific binding was visualized using tetrazolium bromochloroindolylphosphate/nitro blue tetrazolium solution.
Immunocytochemistry
Uninfected and MeV-infected SiHa cells were grown in 35 mm glass-coated wells (Iwaki, Chiba, Japan) and fixed with 4% paraformaldehyde at room temperature for 15 min. After rinsing in PBS, the cells were incubated in PBS containing 0.1% Triton X-100 for 5 min. After rinsing in PBS, the cells were incubated with a blocking buffer (PBS containing 2% BSA and 0.02% Triton X-100) for 20 min. After rinsing in PBS, the cells were incubated for 1 h at room temperature with anti-G3BP1, anti-V, or anti-C protein antibody, and 4′,6-diamidino-2-phenylindole (DAPI). After rinsing in PBS, cells were incubated with Alexa Fluor 488 (green)-conjugated anti-rabbit IgG or Alexa Fluor 594 (red)-conjugated anti-mouse IgG (Life Technologies) for 1 h at room temperature.
After rinsing again in PBS, the cells were embedded with 2% 1,4-diazabicycle [2.2.2.] octane (Sigma-Aldrich, St. Louis, MO) and 50% glycerol in PBS. Specimens were examined and photographed with a ConfoCor3LSM510META microscope (Carl Zeiss, Oberkochen, Germany). Phase-contrast photomicrographs were taken with an OLYMPUS IX71 (Olympus, Tokyo, Japan).
Plaque-forming assay
Vero and Vero/hSLAM cells were cultured in 24-well plates for 3 or 4 days. Supernatants from MeV-infected SiHa cells were centrifuged at 2,000 rpm for 5 min and diluted with 10-fold serial dilutions. The supernatants were inoculated onto Vero or Vero/hSLAM cell for 1 h at 37°C. The cells were then washed twice with PBS, and the medium was replaced by 0.5 mL of 1% carboxymethyl cellulose in RPMI-1640 containing 10% FBS. The cells were then cultured for 5–7 days. After the plates were washed with PBS, the remaining cells were fixed and stained with formaldehyde and gentian violet.
Construction and transfection of MeV C protein expression vector
The MeV P gene was amplified using a OneStep RT-PCR Kit and specific primers (forward; 5′-ATGCATCTCGAGGTGGCAGAAGAGCAGGCA-3′; reverse; 5′-ATGCATCTCGAGCTACTTCATTATTATCTTCATCAGCATC-3′, containing XhoI restriction site). To express the MeV C protein, the start codon sequence in the forward primer was changed to GTG from ATG. Amplified cDNAs were ligated into the XhoI-digested pEB Multi-Puro vector (Wako Pure Chemical). Cells were then transfected with the MeV C protein expression vectors using a Lipofectamine 3000 (Life Technologies), according to the manufacturer's instructions. Forty-eight hours after transfection, the cells were selected with RPMI-1640 containing 10% FBS and 1 μg/mL puromycin.
Results
Establishment of cell lines infected with MeV strains
Four wild strains and one laboratory strain of MeV were used. The genotypes of the three wild strains, MVs/Hokkaido.JPN/52.07/11 [D5], MVi/Hokkaido.JPN/25.11 [D8], and MVs/Sapporo.JPN/19.10/1 [H1], have already been determined (12,14,15). The other wild strain AK1 belonged to genotype D3 (Fig. 1). The laboratory strain Edmonston belongs to genotype A.
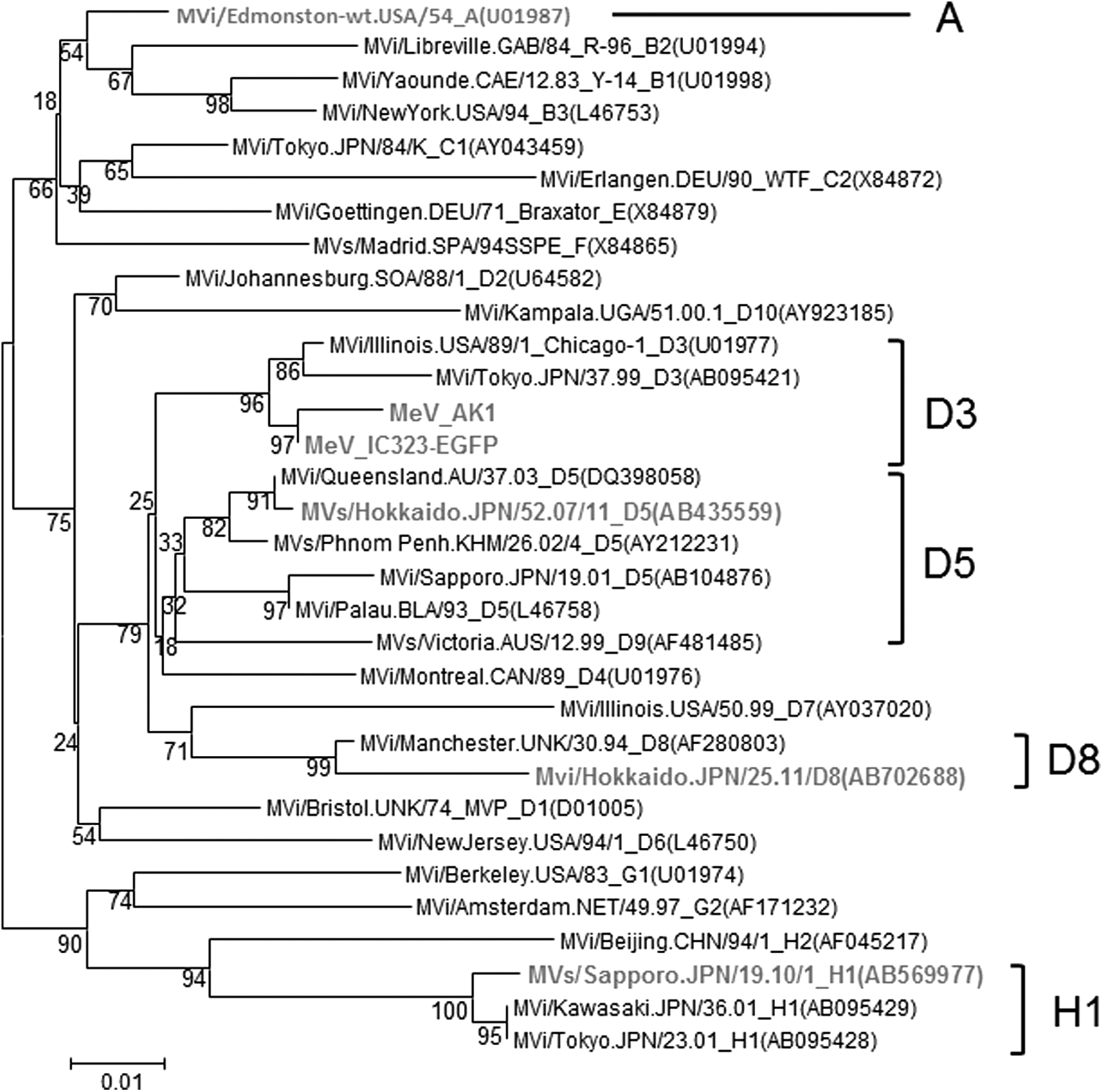
Phylogenetic tree of MeV strains based on the nucleocapsid protein (N) gene sequence. Strains used in this study are expressed in gray letters. MeV, measles virus.
We then established SiHa cell lines persistently infected with these strains, named SiHa-A, SiHa-D3, SiHa-D5, SiHa-D8, and SiHa-H1, respectively. While SiHa cells did not express known receptors for MeV wild strains, namely SLAM and nectin 4 (data not shown), they were susceptible to infection by the MeV wild strains. Furthermore, SiHa cells with no CPE were observed after viral infection. Cells were infected with the different virus strains at a MOI of 0.02. The infected cells were then passaged every 3–4 days. Infection efficacy was monitored using GFP-expressing recombinant MeV (IC323-EGFP). Almost all cells were infected after about 45 days of culture (data not shown). However, SiHa cells infected with Edmonston showed CPE; therefore, we expanded the surviving cell populations.
The presence of MeV N and H genes in the infected SiHa cell lines was confirmed by RT-PCR at 50 days postinfection (data not shown).
We next measured the production of infectious viral particles by MeV-infected SiHa cells during a 4-day cultivation period. All cell lines produced infectious viral particles; however, the virus titers were variable (Table 1). SiHa-D3 produced the highest titer (∼106 PFU/mL) and SiHa-A did the lowest (∼102 PFU/mL). These results confirm the establishment of persistently MeV-infected SiHa cell lines.
Data are expressed as the mean ± standard deviation of three independent measurements.
** p < 0.01 and * p < 0.05, versus virus titer of SiHa-A.
MeV, measles virus; ns, not significant.
Viral protein expression of MeV-infected cell lines
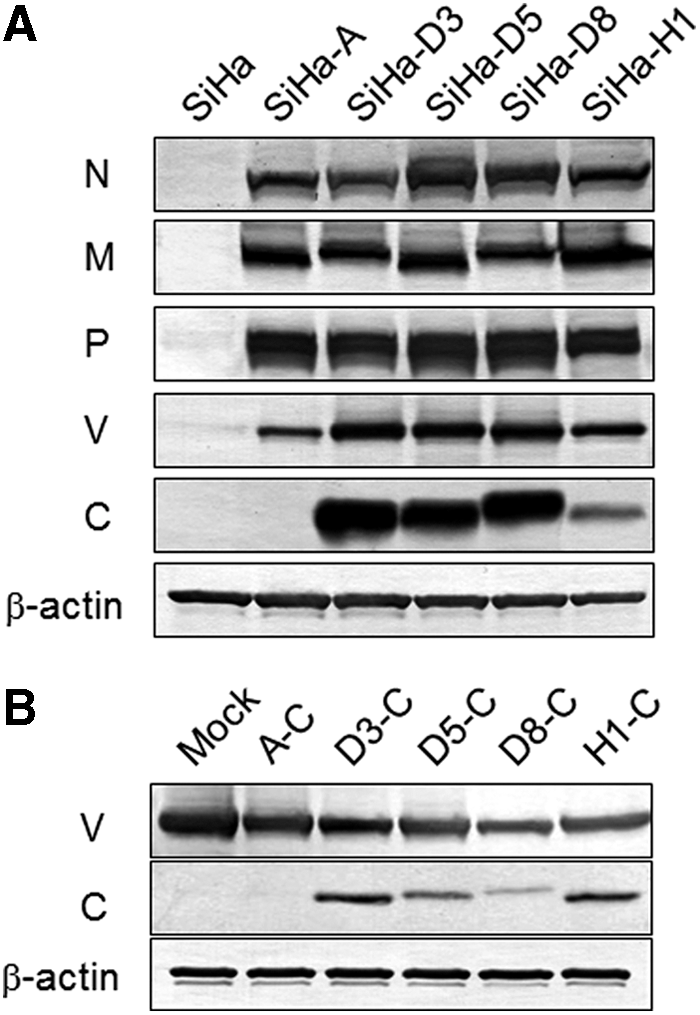
We examined MeV protein expression in MeV-infected SiHa cells (Fig. 2A). Expression of N, M, P, and V proteins was detected in all MeV-infected SiHa cells. However, expression levels of the C protein were variable among the cell lines. SiHa-D3, SiHa-D5, and SiHa-D8 showed strong expression, whereas SiHa-H1 showed a weak expression. C protein was not detectable in SiHa-A cells. The antibody against C protein reacts with both laboratory strain ones and wild stain ones in a similar extent (31).
To determine the amino acid sequence of the C protein, each viral genome was purified from the supernatant of MeV-infected Vero-SLAM, the P gene, which also encodes V and C proteins, was amplified by RT-PCR, and the obtained cDNAs were sequenced. Numerous amino acid alternations were observed in the different strains (Fig. 3). Characteristic amino acid changes on L25P and G44R were observed in wild strains when compared with laboratory strains. R23G, D37G, L85F, S93P, Q96R, N125D, V130M, and T143I were observed in genotype H1 when compared with genotype D strains and laboratory strains.

Comparison of amino acid sequences of C proteins from different MeV strains. White boxes indicate different amino acid residue from other strain's one. Gray boxes indicate different, but the same class amino acid residue from other strain's one.
Expression plasmids for each MeV C protein were transfected into SiHa-A cells and expressing clones were selected with puromycin. SiHa-A cells, whose C protein was not detectable, expressed C proteins from wild strains; however, the expression levels in the transfectants were lower than those in cells infected with the respective MeVs (Fig. 2B). This indicates that SiHa-A cells were able to express C protein. No Edmonston C protein was detected in SiHa-A cells, even after transfection of an expression plasmid harboring cDNA encoding Edmonston C protein.
We next examined the cellular localization of the MeV V and C proteins by immunofluorescence microscopy (Fig. 4). V proteins were detected in all MeV-infected SiHa cells, and were mainly localized in the cytosol and around the cell membrane. C proteins were observed in D3-, D5-, and D8-infected SiHa cells, in which they formed granule-like structures; however, no C protein was observed in H1- and A-infected SiHa cells. The expression levels of these proteins correlated well with the Western blotting results shown in Figure 2A.
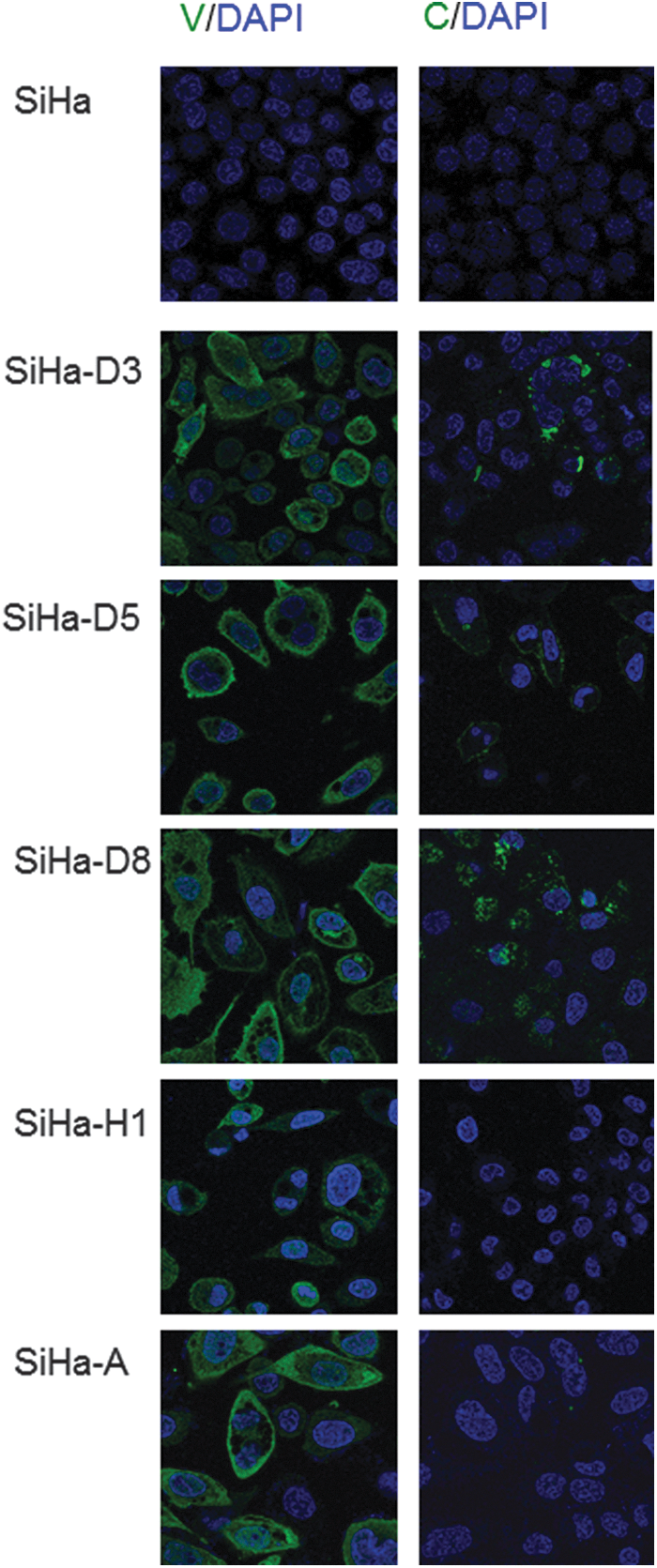
Immunofluorescence microscopic observation of MeV V and C protein expression in MeV-infected and uninfected SiHa cell. Left panels: Cells were stained with an anti-MeV V protein antibody. Right panels: Cells were stained with an anti-MeV C protein antibody. The specimens were visualized with an Alexa Fluor 488-labeled secondary antibody (green) for the binding of the primary antibody and nuclei stain with DAPI (blue). DAPI, 4′,6-diamidino-2-phenylindole.
Previous studies show that several stresses, including chemicals and virus infection, induce the formation of granule structures, termed stress granules (SGs), in the cytosol (19,20). Therefore, we examined whether the C-protein-containing granules were in fact SGs. In a control experiment, we confirmed whether SiHa cells formed SGs containing a well-established maker of SGs, G3BP1, after the treatment with 0.5 mM sodium arsenite for 30 min (Fig. 5A). The C protein-containing granule-like structures within SiHa cells infected with D3, D5, and D8 did not colocalize with G3BP1 (Fig. 5B); therefore, these granules were not SGs.

Immunofluorescence microscopic observation of localization of G3BP1 protein (a marker of SGs) and MeV C protein in MeV-infected SiHa cells.
DI particle genome was not detected in MeV-infected SiHa cells
Cells persistently infected with MeV produce DI particles, which interfere with IFN signaling (22,24). Therefore, we examined MeV-infected SiHa cells for the presence of DI particles by the detection of RNA copy back using two previously established RT-PCR methods (4,25,30). No RNA derived from DI particles was detected in infected SiHa cells or their culture supernatants (data not shown).
MeV-infected cell lines showed altered ISG expression
We measured the mRNA expression of several ISGs [MxA, 2′-5′-OAS, and IRF-1] in MeV-infected and uninfected SiHa cells by real-time RT-PCR (Tables 2 –4, respectively). All ISGs examined were upregulated upon MeV infection compared with uninfected SiHa cell. The mRNA expression of MxA and 2′-5′-OAS was mostly inhibited by the Jak inhibitor, ruxolitinib. The mRNA expression of IRF-1 was partially (∼33%) inhibited by ruxolitinib. Upregulation of these genes, notably MxA mRNA, varied among the different infected MeV strains. The ability of MeV to upregulate these genes was higher in the order of A>H1>D8>D5>D3. The upregulation levels were correlated with suppressed amounts of virus particle production.
The mRNA levels were normalized against those of β-actin. Values are expressed relative to that of untreated/uninfected SiHa cells, which were set at 1.00. Data are expressed as the mean ± standard deviation of three independent measurements. Values in parenthesis indicate average values for untreated cells.
‡ p < 0.01 and †p < 0.05, versus value of SiHa cell.
Values represent the ratio of the mRNA level between untreated and ruxolitinib-treated cells.
** p < 0.01 and * p < 0.05, versus each untreated cell.
IFN, Interferon; Jak, janus kinase.
The mRNA levels were normalized against those of β-actin. Values are expressed relative to that of untreated/uninfected SiHa cells, which were set at 1.00. Data are expressed as the mean ± standard deviation of three independent measurements. Values in parenthesis indicate average values for untreated cells.
‡ p < 0.01 and †p < 0.05, versus value of SiHa cell.
Values represent the ratio of the mRNA level between untreated and ruxolitinib-treated cells.
* p < 0.05, versus each untreated cell.
The mRNA levels were normalized against those of β-actin. Values are expressed relative to that of untreated/uninfected SiHa cells, which were set at 1.00. Data are expressed as the mean ± standard deviation of three independent measurements. Values in parenthesis indicate average values for untreated cells.
‡ p < 0.01 and †p < 0.05, versus value of SiHa cell.
Values represent the ratio of the mRNA level between untreated and ruxolitinib-treated cells.
** p < 0.01 and * p < 0.05, versus each untreated cell.
We also analyzed the effect of IFNs [type I (IFN-α), II (IFN-γ), and III (IFN-λ)] on MeV-infected SiHa cell lines (Tables 2 –4). Both IFN-α and -λ strongly induced MxA mRNA expression in uninfected SiHa cells (254- and 104.8-fold, respectively) after 8 h of treatment, respectively (Table 2). However, induction of MxA mRNA induced by all IFNs abolished in all MeV-infected SiHa cells. IFN-γ treatment for 8 h led to weaker induction of MxA mRNA (12.1-fold) in uninfected SiHa cells. Similarly, expression of 2′-5′-OAS mRNA was induced in uninfected SiHa cells after IFN treatments (35.8-, 14.1-, and 4.9-fold higher expression after treatment with IFN-α, -λ, and -γ, respectively; Table 3). In contrast, no significant IFN-mediated induction of 2′-5′-OAS mRNA was observed in all MeV-infected SiHa cells. IFN-γ, but not IFN-α and -λ, induced the expression of IRF-1 in the infected SiHa cells (Table 4). IFN-γ also induced IRF-1 mRNA expression in all MeV-infected SiHa cells; however, the levels were much less than those in uninfected SiHa cells. The levels in SiHa-D3 cells were decreased one-eighth, and the levels in the other MeV-infected SiHa cells one-third to one-half, than that in uninfected SiHa cells.
IFN production by MeV-infected SiHa cells
MeV-infected SiHa cells, but not by uninfected cells, produced IFN-β and IFN-λs (Table 5). The level of IFN productions followed the following order: A>D3>D5>D8>H1. Only very low levels of IFN-α were detected in SiHa-A cells. Treatment with ruxolitinib indicated that IFN-β production was completely Jak independent, whereas that of IFN-α was Jak dependent. IFN-λ production was partially Jak dependent. No IFN-γ was detected in the culture supernatants from all MeV-infected and uninfected SiHa cells.
nd, not detected.
Effect of ruxolitinib on viral particle production
We examined whether ruxolitinib affected virus production by MeV-infected SiHa cells (Table 1). Ruxolitinib led to significant suppression of infectious virus production by SiHa-A cells. However, virus particle production by wild strain-infected SiHa cells was not affected by treatment with ruxolitinib. Ruxolitinib-mediated inhibition of the Jak pathway was confirmed by suppressed STAT1 phosphorylation and reduced MxA protein expression (Fig. 6; lower panels). Ruxolitinib did not alter the expression levels of viral proteins (P, C, and V) in the infected cells (Fig. 6; upper panels).
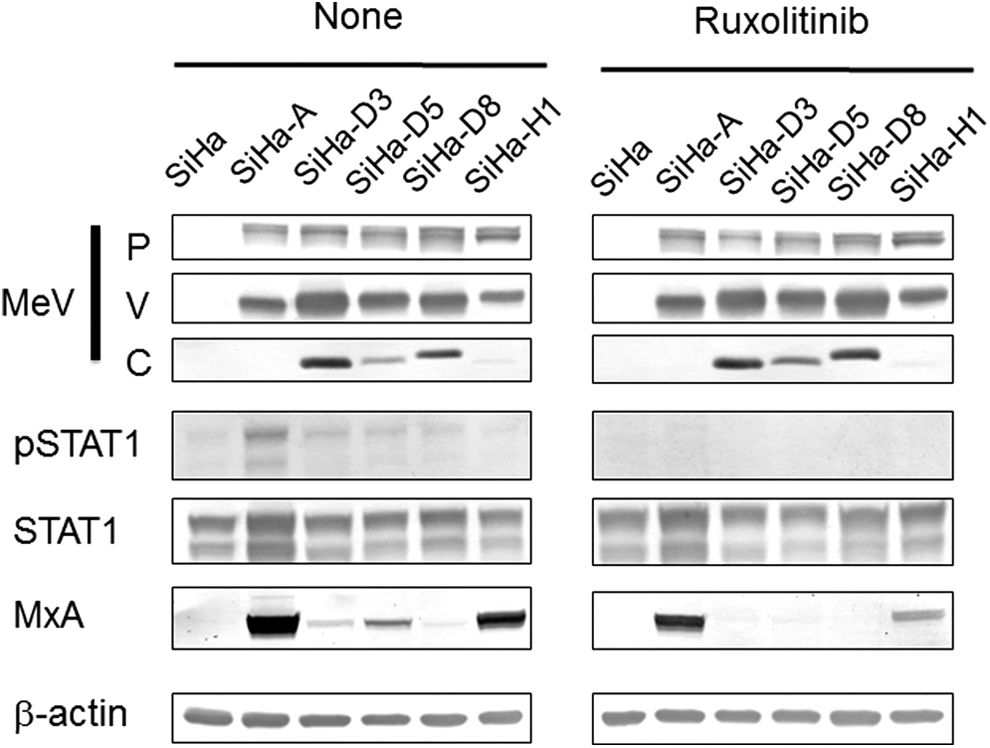
Western blot analysis of viral protein, STAT1, Tyr-phosphorylated STAT1 (pSTAT1), and MxA expression in uninfected SiHa cell and MeV-infected SiHa cells in the presence or absence of ruxolitinib. β-actin was used as a control.
Discussion
MeV suppresses the type I IFN signaling pathway to counteract the host innate immune response. Previous studies show that immune suppression is mediated by the MeV V protein (2,3,18,27). We also observed suppression of IFN system in SiHa cell lines persistently infected with MeV, although type II IFN (IFN-γ) signaling is not suppressed (33). Furthermore, IFN-γ-like signaling, such as phosphorylation of STAT1 and Jak1, are constitutively activated in SiHa cells infected with MeV laboratory strains (31,32). Suppression of type I IFN signaling is observed in cells infected with all MeV strains, including laboratory and wild strains, so far examined, although the IFN-γ-like signals are transduced less well in cells infected by wild strains than those by laboratory strains.
However, previous studies examined only one genotype, D3, as a wild strain. The aim of this study was to examine whether MeV wild strains of different genotypes commonly share the ability to suppress IFN-related antiviral systems in the host.
We examined the effect of MeV wild strains belonging to genotypes D5, D8, H1, and AK1 [D3] and the laboratory strain Edmonston [A] on ISG expression and virus particle production by infected cells. First, we established persistently MeV-infected SiHa cell lines. Although SiHa cells were negative for known receptors for MeV wild strains, namely SLAM and nectin 4 (5,17), they were susceptible to infection by MeV wild strains. Also, the wild strains, but not the laboratory strains, did not cause CPE. However, the mechanism of infection remains unclear. Repetitive passage after infection led to the establishment of persistently infected SiHa cell lines. The mode of infection was different for wild and laboratory strains.
ISG expression can divide IFN-induced (IFN-dependent) and MeV infection-induced (IFN-independent). MeV infection upregulated ISG expression, although we found that the levels of upregulation varied among strains. Edmonston induced higher upregulation than wild strains. Type I and III IFN-induced ISG expression was almost completely suppressed in all MeV-infected SiHa cells, suggesting that ISG expression was in an IFN-independent manner. Also, IFN-independent ISG expression was induction in a Jak-dependent manner.
SiHa cells and MeV-infected SiHa cells did not produce IFN-γ, whereas IFN-α, β, and λ were produced by SiHa-A cells. Production of IFN-β was Jak independent, that of IFN-α, which was detectable only in SiHa-A cells, was Jak dependent, and that of IFN-λ was partially Jak dependent. The results indicate that MeV genotype A was a strong inducer of IFN-independent ISG expression and Jak-independent production of IFN-β and λs. IFN-independent ISG expression inducer upon MeV infection followed the order A>H1>D8>D5>D3 (Tables 2 –4). This order was inversely correlated with the expression levels of MeV C protein (Fig. 3A).
Our previous studies (31,32) indicate that MeV potentially upregulates IFN-γ-like signaling and expression of IRF-1. However, the C protein interacts with phosphorylated STAT1, suppresses STAT1 dimer formation, and inhibits MeV-induced IFN-γ-like signaling. SiHa cells infected with wild strains expressed C protein at a markedly higher level than cells infected with laboratory strains.
Lines of evidence suggest that SiHa cells infected with wild strains showed downregulated MeV-induced ISG expression, which might be related to C protein levels. While MeV-induced ISG expression varied among strains, MeV almost completely suppressed type I and type III-induced ISG expression in all transfected cell lines. However, MeV only partially inhibited IFN-γ-induced ISG expression.
Cells persistently infected with MeV are known to produce DI particles, which interfere with IFN signaling (22,24). Shingai et al. reported that 5′-copyback DI is found in laboratory strain-infected cells (22,23). Pfaller et al. showed that C protein impairs DI particle production (21). These findings suggest that differences in IFN-related signal transduction between genotype A (laboratory strains) and the other genotypes (wild strains) may be due to DI production by genotype A. However, we did not observe expression of DI particle genes, namely 5′-copyback, in any of the MeV-infected SiHa cells examined in this study.
We also examined the cellular localization of MeV C and V proteins (Fig. 4). The V protein localizes in the cytosol, particularly around cell membrane. This might correspond with our previous results showing that the V protein interacts with the IFN-α receptor complex, and therefore suppresses signal transduction from the receptor, such as failure of STAT1 recruitment (33).
The C protein localized in the cytosol where it formed granule-like structures. These granule-like structures were unrelated to SGs, which are induced by viral infection and by double stranded RNA (20). A previous study showed that MeV suppresses SG formation by inhibiting PKR, which is induced by C-protein-mediated activation of RNA-specific adenosine deaminase 1 (ADAR1) during infection (19). The granule-like structures containing the C protein have not been characterized; however, they may sequester signal transduction molecules related to IFN signaling. It seems to be agreed that the protein C contributes to efficient viral replication (1,6).
However, the mode of expression and function of the C protein are highly controversial. Nakatsu et al. (16) suggest that the C protein in Vero/hSLAM cells infected with MeV expressed as a granule-like structure in the cytosol, similar to the findings in this study. However, the expression levels of the C protein and its ability to recover virus replication are similar for the wild strain and the Edmonston strain. They concluded that both types of MeV harbor a fully functional C protein. Ito et al. (10) also showed granule-like expression of the C protein in the cytosol using a MeV minigenome system. They suggest that the C protein colocalizes with host Shc Src homology two domain-binding protein 1 (SHCBP1), which is required for the viral RNA polymerase activity.
Sparrer et al. (26) showed that wild strain C proteins share a nuclear localization signal and localize in the nuclei, whereas C protein from laboratory strains are dispersed throughout the cytosol. The nuclear-localized C protein suppresses IFN-γ transcription. Therefore, they concluded that the absence of a nuclear localization signal from the laboratory strain C protein is important for attenuation of MeV. However, they used 293T cells and HeLa cells transfected with C protein expression plasmids, but did not perform virus infection experiments. This study found that different MeV strains expressed different levels of C proteins.
The reasons for the contradiction remain unclear. One reason may be differences between experiment systems.
In conclusion, suppression of both IFN-induced and MeV infection-induced antiviral immunity are required for efficient viral replication. The suppressive activity of MeV was varied among strains and that of genotype D wild strains was greater compared with laboratory and genotype H1 strains, which may be a reason for the high virulence of wild strains. In other words, lack of suppression of MeV-induced antiviral activity might attenuate laboratory strains. Furthermore, the ability to suppress MeV infection-induced antiviral response varied among wild strains with different genotypes.
Footnotes
Acknowledgments
The authors thank Drs. Setsuko Ishida, Masahiro Miyoshi, Rika Komagome, and Motohiko Okano (Hokkaido Institute of Public Health) for providing the MeV wild strains.
This work was partly supported by Grants-in-Aid for Scientific Research from Japan Society for the Promotion of Science (No. 26293370 and No. 26670746) (T.H.), and by a GSK Japan Research Grand 2015 (N.O).
Author Disclosure Statement
No competing financial interests exist.