
Abstract
Zika virus (Zika V) is a positive single-stranded RNA virus that is transmitted by mosquito bites. Zika V Envelop protein is antigenic and is involved in fusion and entry of viral particles into the cell. Till date, there is no vaccine and antiviral drug available against Zika V. Thus, there is a need to develop a vaccine against Zika V. This study was designed for the prediction of B cell and T cell epitopes that can be helpful in diagnosis and vaccine designing against this emerging threat. For this purpose, several B cell and T cell epitopes were predicted that are conserved among Zika virus genomes taken from 12 different countries. Peptides QTLTPVGRL, in case of major histocompatibility complex (MHC) class I, and IRCIGVSNRDFV, in case of MHC class II, are highly antigenic among T cell epitopes. Molecular docking was performed to study the interactions of B cell epitopes with HLA-B7. However, these predicted epitopes could play a constructive role in designing of a vaccine against Zika V.
Introduction
Z
Zika V produces a polyprotein 5′-C-prM-E-NS1-NS2A-NS2BNS3-NS4A-NS4B-NS5-3′ that on cleavage gives Envelop protein (E), capsid (C), precursor of membrane (prM), and seven different nonstructural (NS) proteins (16). Among structural proteins, Zika Envelop protein is involved in binding, fusion, and entry of virus into host cells (17). So, E protein can serve as a target to stop or block the entry of the virus into host cells.
For purposeful designing of peptide vaccine, it is considered necessary to find out the epitopic regions in immunogenic protein, such as Envelop protein in this case. Immunoinformatics is a computer-based modeling, representation, analysis, and study of immunological data (3). Recognition of antigens at a molecular level helps in the development of peptide vaccines. The basic idea behind this strategy is based on chemical methods to synthesize B and T cell epitopes that can induce a specific immune response. For the first time, in 1985, a peptide-based vaccine was synthesized (14). Immunoinformatics mostly focuses on the study and designing of algorithms that are utilized for in silico prediction of B and T cell epitopes that helps in reducing the cost as well as time. Using these tools, one can predict the areas with potential binding sites within the sequence (20). Analysis of the genome of pathogen to find out the antigenic proteins is called reverse vaccinology (21).
In silico peptide prediction has been used in several cases such as in silico peptide development against Chikungunya (12) virus and hepatitis C virus (13). Therefore, the present study was designed and several tools of Bioinformatics and immunoinformatics were used for the prediction of conserved B and T cell epitopes among genomic sequences of Zika V Envelop protein.
Methodology
Sequence retrieval and primary structure analysis
Sequence of the Zika V Envelop protein was taken from Genbank using the accession number (AIC06934.1). Protparam (11) is a freely available online tool that is used for analysis of primary structure of the protein. Different parameters such as grand average of hydropaticity (GRAVY), amino acid composition, estimated half life, atomic composition, stability index, and molecular weight were calculated. JPred (6) and Psipred (2) online servers were used for prediction and analysis of secondary structure of the protein.
Three-dimensional structure retrieval
Three-dimensional (3D) structure of the target protein was retrieved from Protein Data Bank (PDB), using 5ire as PDB_ID. This structure was determined through the electron microscopy technique with resolution 3.8 Å.
Prediction of B and T cell epitopes
B and T cell epitopes of the Zika V Envelop protein were designed using a systematic strategy. Antigenicity of the Envelop protein was calculated by an online tool Vaxijen 2.0 (5). TMHMM was used for analysis of transmembrane topology of the E protein (15). BCPREDS is a freely accessible online server that is used for the prediction of B cell epitopes (7). Only those epitopes were selected for further analysis that were part of the exposed region and were not part of the transmembrane region. Vaxijen 2.0 was again used for the antigenicity calculation of selected epitopes. DiscoTope 2.0 was used for the prediction of B cell epitopes from 3D structure of the Zika V Envelop protein. Furthermore, positional confirmation of predicted epitopes was done by an online tool EpiSearch. In addition to that, T cell epitopes were also predicted. Propred-1 and Propred were used for screening of MHC class I and MHC class II alleles, respectively. Both tools contain a large number of alleles of human leukocyte antigens (HLAs); therefore, their prediction is acceptable. Immunoproteasome and proteasome filters were set to 5% for the class I alleles.
Conservation analysis of selected epitopes
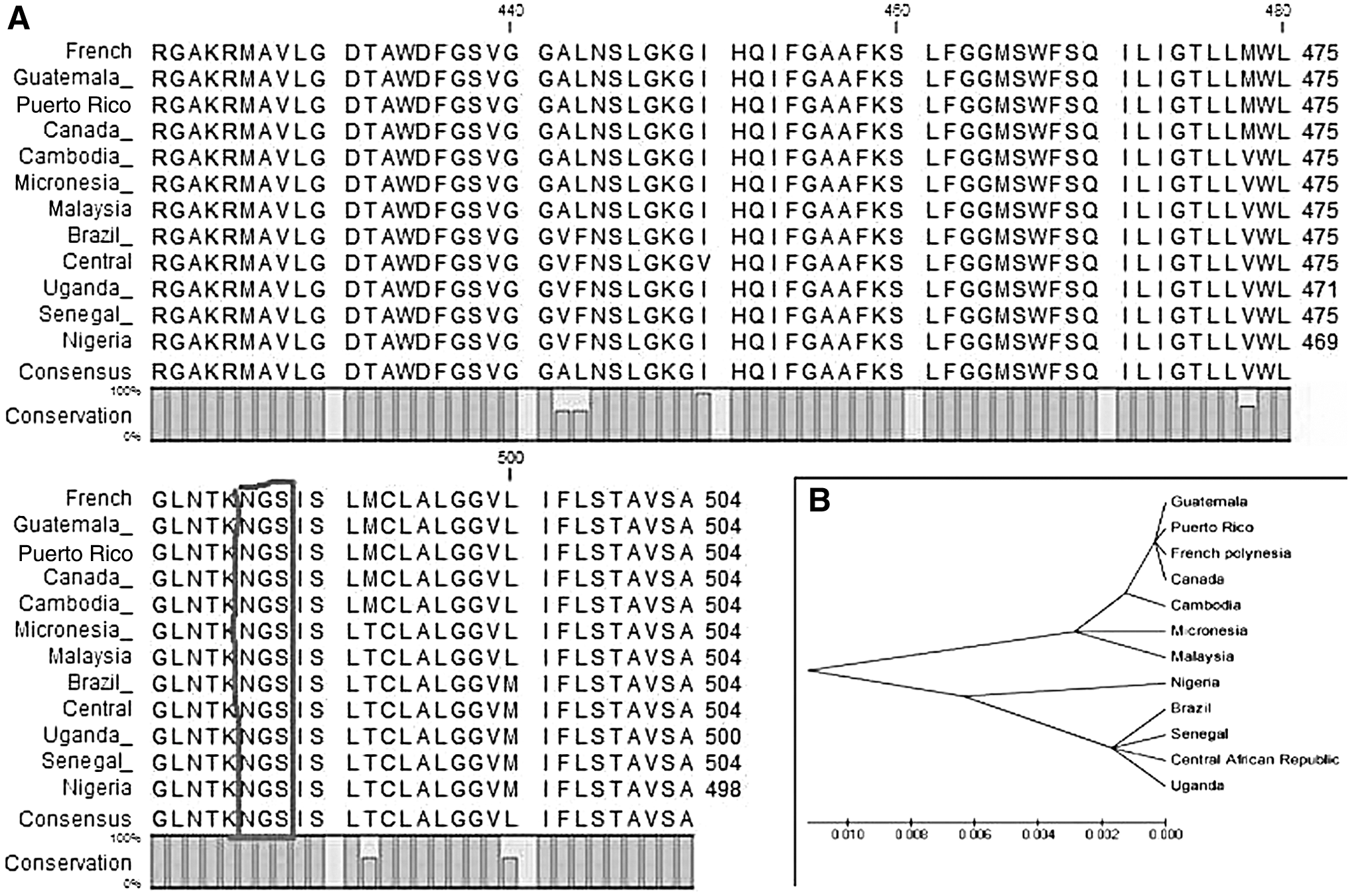
Sequences of the Zika V Envelop protein that belong to 12 countries were retrieved from Genbank; a consensus sequence was developed for each country, which was used further for multiple sequence alignment to analyze the conservation of different epitopes. A phylogenetic tree was also generated through this alignment as input. Only those epitopes that were present in the conserved region were selected for further analyses. Furthermore, conservation of selected epitopes was checked through IEDB (22).
Molecular docking
Molecular docking of the predicted epitopes was performed with their respective alleles to study their interactions and binding with alleles. The 3D structure of three epitopes was predicted using the PEP FOLD at RPBS MOBYL portal (9,18). The 3D structure of the HLA-B7 allele was taken from PDB through its PDB_ID: 3VCL. It was determined by the X-ray diffraction method, and its resolution was 1.7 Å. The structure of HLA-B7 was already bound with a peptide RPHERNGFTVL. The already bounded peptide was removed, and the same pocket was selected for the docking of newly predicted epitopes to study their interactions with the receptor. Energy of receptor (HLA-B7) was minimized before docking. Molecular operating environment (MOE) was used for docking (4). For calculation of scores, London DG tool of MOE was used and refinement was performed by applying force field. Ten confirmations were allowed for each epitope.
Allergenicity assessment
Aller Hunter online available tool was used for allergenicity assessment of predicted epitopes. It compares the input sequence with already reported allergens. It predicts allergens and nonallergens very accurately; therefore, it is used for allergenicity checking of proposed peptides.
Results
Model description
A sequence of Zika V Envelop protein was taken from Genbank using AIC06934.1 as an accession number. Results of protein primary structure analysis showed that the theoretical isoelectric point (PI) of protein is 6.51 and its molecular weight is 54380.1 Da. It was predicted as a negatively charged protein, because its isoelectric point was below seven. Totally, 51 amino acids were considered as negatively charged and 47 were positively charged. The instability index of protein was 22.77, which showed that the protein is stable. I (Ile) was considered as an N-terminal residue of the target protein. The calculated Aliphatic index of protein was 81.45, and GRAVY was −0.083. Secondary structure analysis showed that protein contains 15% alpha helix, 50% beta strands, and 24% coils (Fig. 1A). Furthermore, DiANNA 1.1 was used for prediction of disulfide (S–S) bonds present in target protein (9). Totally, 13 cysteine residues that were found in whole sequence and six disulfide bonds were predicted at positions 3–105, 30–116, 60–92, 74–121, 190–308, and 339–488.

represents alpha helix, and  represents beta sheets.
represents beta sheets.
The 3D structure of protein is crucial to understand and analyze functions, interactions, and localization of protein. The 3D structure of the protein was taken from PDB, which was determined via electron microscopy (Fig. 1B, C).
Conservation analysis
Sequences of Zika V Envelop protein from 12 different countries (maximum three sequences from each country) were taken, and a consensus sequence was drawn using CLC workbench, an offline tool. For the conservation analysis, consensus sequences were subjected to multiple sequence alignment through ClustalW and a phylogenetic tree was also constructed based on this alignment via MEGA 5.0 (Fig. 2B). A glycosylation site was also searched with Asn-X-Ser as a formula, whereas X represents any amino acid. A conserved N-glycosylation site was found at position 486. It was conserved among all the sequences of different countries (Fig. 2A).
B and T cell epitope prediction
The antigenicity of target protein was 0.5444, and it was calculated by an online server Vaxijen 2.0, which represented that it was antigenic. The TMHMM online freely accessible tool was used for the prediction of transmembrane topology of the target protein, which showed that there were two transmembrane helixes that range from 455 to 477 and from 484 to 503. Residues 1–454 were exposed outside, residues 483–503 were buried inside, and residue 504 was present at the surface.
Prediction of B cell epitopes
B cell epitopes play an immense role in activation of the immune system and prevention from viral infections. The BCPRED online server was used for the prediction of B cell epitopes, with 75% specificity. The length of epitopes was sufficient to maintain 14 residues. Many epitopes were predicted by BCPRED but only eight epitopes were antigenic, which are shown in Table 1. Epitope LMWLGLNTKNGSIS with a score of 0.879 and an antigenicity of 1.8495 was predicted as part of a transmembrane helix; therefore, it could not act as an antigen and is marked in bold in Table 1. Epitopes SRCPTQGEAYLDKQ, VDRGWGNGCGLFGK, LELDPPFGDSYIVI, LGDTAWDFGSVGGA, LEHGGCV TVMAQDK, DTGHETDENRAKVE, and LGLDCEPRTGLDFS with 0.984, 0.954, 0.926, 0.9, 0.86, 0.855, and 0.778 scores, respectively, are part of exposed regions and have the ability to induce an immune response.
Boldface epitopes don't act as antigen because they are present in transmembrance helix.
Scores and antigenicity of epitopes showed that these peptides can play an important role in the initiation of immune responses. In addition to that, Disco Top 2.0 server was used for prediction of B cell epitopes from 3D structure. The threshold was set at −2.0, and it calculated a total of seven epitopes in different exposed regions. The peptide starting from position 279 (GLY) has not shown any contact residue, therefore it was excluded (Table 2). The positions of these predicted epitopes are marked as yellow in 3D structure (Fig. 3). Furthermore, conformational analysis was also done via an online tool EpiSearch. Episearch confirmed the position of predicted epitopes by using the 3D structure and peptide sequence (Fig. 4).
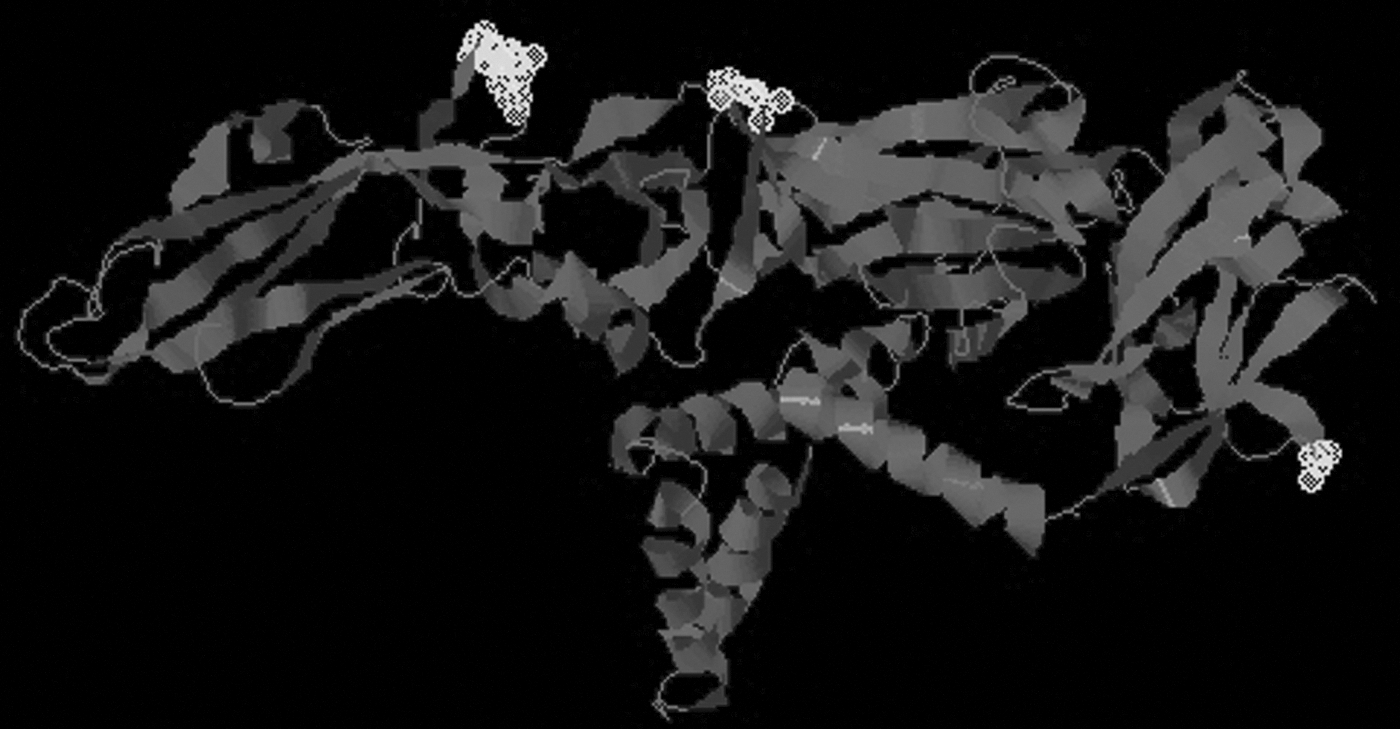
Discotop prediction by using 3D structure. Epitope positions are marked as gray.
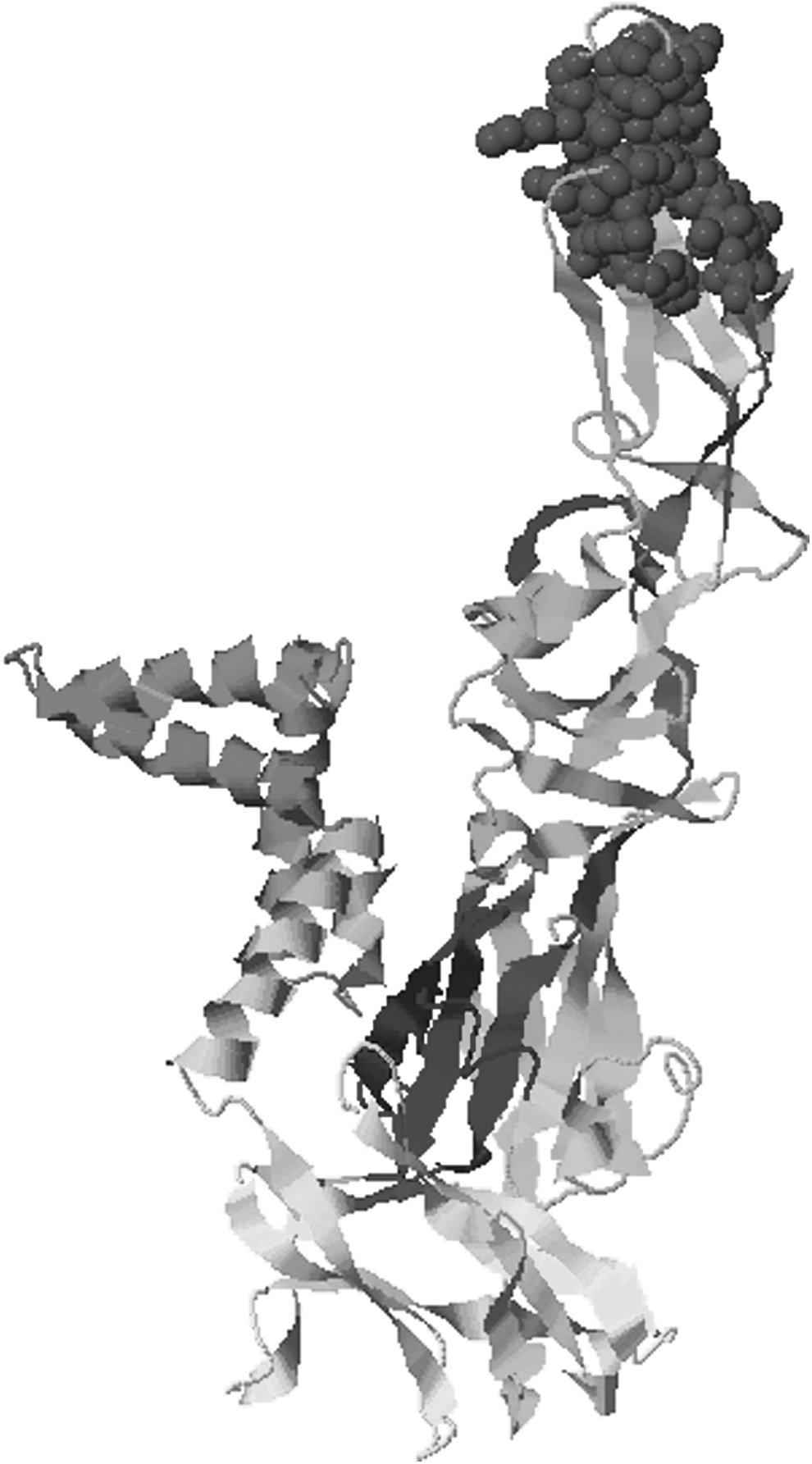
Conformational analysis of predicted epitope through Episearch.
Prediction of T cell epitopes
Prediction of T cell epitopes of target protein was done by two different online servers. Prediction of MHC class I alleles was done using Propred-I. Propred was used for MHC class II alleles. ProPred1 is a freely accessible tool that is used for prediction of peptides that bind to MHC class I alleles. The sequence of E protein of the Zika V was given as an input in FASTA format, the threshold was set at 4%, and immunoproteasome and proteasome filters were kept on. Different peptides against all MHC class I alleles were predicted, but only antigenic peptides were screened through antigenicity testing. Antigenicity of “QTLTPVGRL” peptide was calculated as the highest among all peptides (Table 3).
HLA, human leukocyte antigen; MHC, major histocompatibility complex.
Sequence was also given in FASTA format to Propred for prediction of peptides that bind to MHC class II alleles. Antigenicity testing was also performed by using Vaxijen 2.0, and only peptides possessing an antigenic nature were selected for further analysis. Antigenicity of “IRCIGVSNRDFV” peptide was higher as compared with all other MHC class II alleles binding peptides (Table 4). Furthermore, conservation analysis was also performed via IEDB. All the B and T cell epitopes were conserved among sequences of all countries used in the study, except DTGHETDENRAKVE. For Malaysia, the sequence of this peptide was DXGHETDENRAKVE. For Brazil and the Central African Republic, DIGHETDENRAKVE could act as an epitope. This peptide could not act as an epitope in Uganda and Nigeria.
Interactions of some selected epitopes with HLA-B7
3D structures of the four epitopes GGTWVDVVL, NSPRAEATL, AKRQTVVVL, and QTLTPVGRL were predicted by PEP-FOLD. The 3D structure of HLA-B7 was retrieved from PDB, which was already bound with a peptide RPHERNGFTVL. Peptide removal, protonation, and energy minimization were done before docking. 3D structures of these peptides were docked against the receptor. Ten best confirmations of each peptide were calculated. Interacting residues and docking scores of each peptide are given in Table 5. Furthermore, interactions of these peptides were studied against HLA-B7 by creating a molecular surface and without a molecular surface (Fig. 5). The highest score (−22.43) containing peptide was AKRQTVVVL, which showed interactions with Ser 61 (B chain), Tyr 63 (B chain), Ala211 (A chain), and Thr233 (A chain). Interactions of these peptides with the Zika V Envelop protein are shown in Fig. 5.
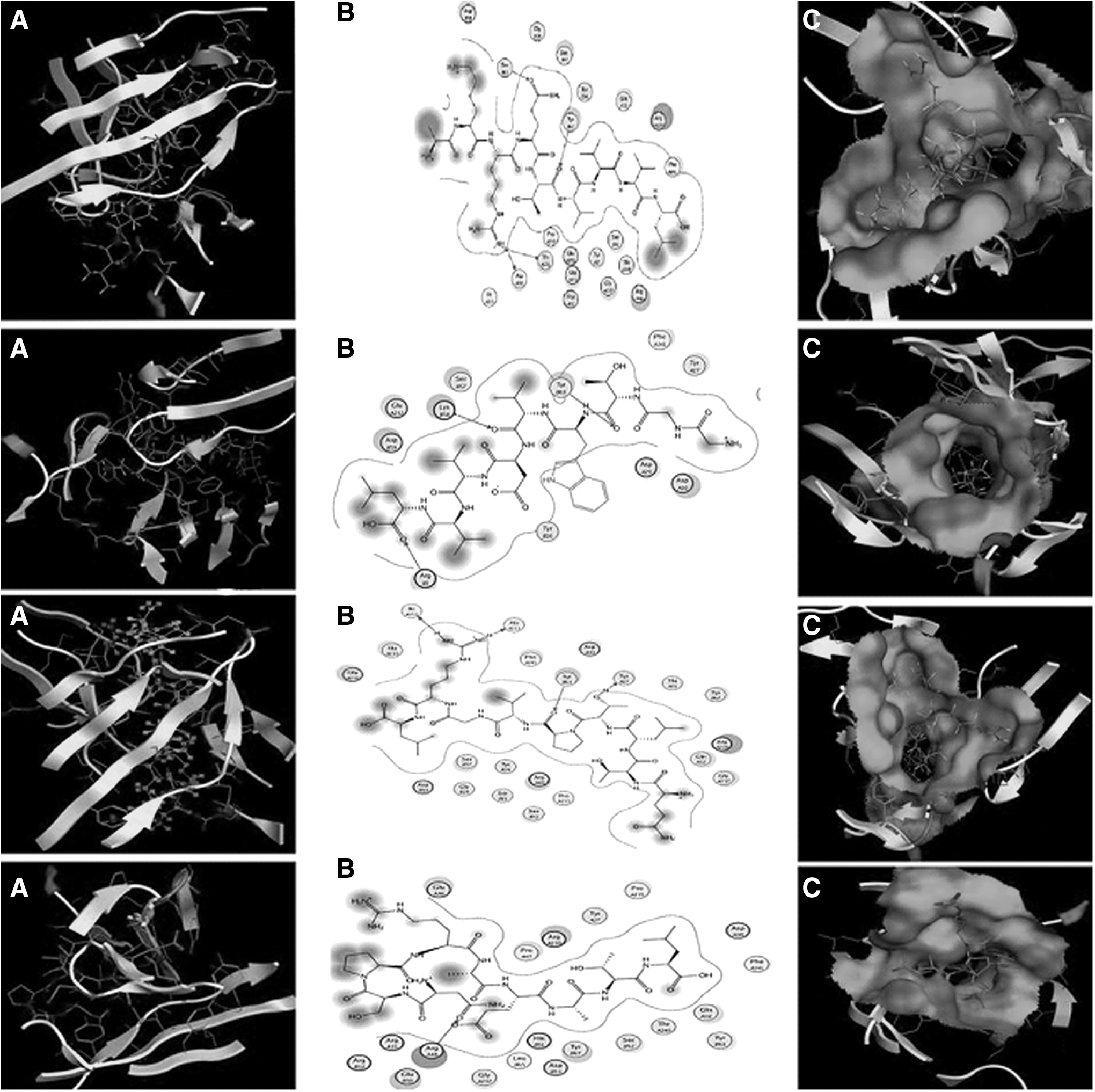
Docking interactions.
Allergen testing
Allergen testing was performed through Aller Hunter. According to FAO/WHO evaluation scheme, query sequence does not fulfill the criteria set for allergens. The query sequence was predicted as a nonallergen, with a score of 0.0 (sensivity (SE) = 91.6%, specificity (SP) = 89.3%).
Discussion
More than 7 billion people are living in this world. A number of diseases that occurred due to different viruses are discovered. The rate of these diseases is higher in developing countries. So there is a dire need to cope with these devastating diseases. Medical biotechnology plays a very important role in controlling the pace of this disaster. In addition to it, bioinformatics, immunoformatics, and chemoinformatics are playing very important roles in decreasing the time and cost in drug and vaccine designing. Zika V is an emerging threat to the world; it is spreading in different countries of Africa and Malaysia.
It exhibits dengue-like symptoms, headache, fatigue, fever, and microcephlly in newborns as well. There is no vaccine available against Zika V. So, there is a dire need to prepare a vaccine or drug that could restrict this disaster. E protein of the Zika V plays an important role in fusion and entry of the Zika V in human cells. Therefore, E protein could be considered a target to restrict the entry of virus into the cell. The focus of the present study is to find out the different B and T cell epitopes that could play important roles in the initiation of immune responses. In this study, primary and secondary structure analysis, 3D structure prediction, and epitope designing were performed. Different tools such as JPred and Psipred were used for analysis of primary and secondary structures. Primary structure analysis revealed that the molecular weight of protein is 54380.1 Da, and protein has 13 cysteine residues and five disulfide bonds. I (Ile) was predicted as the N-terminal residue of protein.
3D structure plays an immense role in understanding the character and functions of protein, which were taken from PDB. It is also very important in protein– protein interaction studies, and it can be used as a target in case of drug designing. The focus of the present study was on prediction of B and T cell epitopes. Antigenicity of the Envelop protein showed that a part of it can act as an initiator of immune response. A total of eight B cell epitopes were predicted; all these epitopes were antigenic and present at the outer surface of protein, except LMWLGLNTKNGSIS, which is part of the transmembrane helix.
Conservation analysis of these predicted epitopes was performed among the genomic sequences of Zika V belonging to different countries. All predicted B cell epitopes were conserved among all genomic sequences of Zika V that were included in the present study, except DTGHETDENRAKVE. Furthermore, the locations of predicted epitopes were analyzed using 3D structure of Envelop protein. MHC class I and MHC class II epitopes were predicted through online servers Propred-I and Propred, respectively. Antigenicity of these predicted epitopes was checked. QTLTPVGRL, in case of MHC class I, and IRCIGVSNRDFV, in case of MHC class II, were found to be highly antigenic, which indicates that these epitopes can play an important role in the initiation of immune responses.
Furthermore, conservation analysis of these epitopes was performed and all T cell epitopes were conserved among all genomic sequences of Zika V belonging to 12 different countries.
Conclusion
For development of effective vaccines, it is crucial to hit the antigenic parts of the virus, thus directing the immune system toward saving the host from viral attack and infection. Therefore, the present study was conducted to determine or predict epitopes (antigenic determinants) of E protein of Zika V. Results of the present study showed that all the predicted epitopes are antigenic, are mostly conserved among 12 countries, and are able to initiate immune responses. These epitopes could be very helpful in the diagnosis of Zika V and also in the development of a vaccine against Zika V to protect the world from this emerging menace.
Footnotes
Author Disclosure Statement
No competing financial interests exist.