
Abstract
Porcine rotavirus-A (PoRVA) is one of the common causes of mild to severe dehydrating diarrhea, leading to losses in weaning and postweaning piglets. A rapid, highly specific, and sensitive antigen-capture enzyme-linked immunosorbent assay (AC-ELISA) was developed for detection of PoRVA, by using VP6 (a highly conserved and antigenic protein of group-A rotavirus)-directed rabbit polyclonal antibodies (capture antibody) and murine monoclonal antibodies (detector antibody). The detection limit of AC-ELISA was found to be equal to that of conventional reverse transcription–polymerase chain reaction (RT-PCR; about 102.5 TCID50/mL). For validation of the in-house AC-ELISA, 295 porcine fecal/diarrhea samples, collected from different provinces of China, were evaluated and compared with conventional RT-PCR and TaqMan RT-quantitative PCR (qPCR). The sensitivity and specificity of this in-house AC-ELISA relative to RT-qPCR were found to be 91.67% and 100%, respectively, with the strong agreement (kappa = 0.972) between these two techniques. Total detection rate with AC-ELISA, conventional RT-PCR, and RT-qPCR were found to be 11.2%, 11.5%, and 12.2%, respectively, without any statistical significant difference. Moreover, AC-ELISA failed to detect any cross-reactivity with porcine epidemic diarrhea virus, transmissible gastroenteritis virus, pseudorabies virus, and porcine circovirus-2. These results suggested that our developed method was rapid, highly specific, and sensitive, which may help in large-scale surveillance, timely detection, and preventive control of rotavirus infection in porcine farms.
Introduction
R
Rotaviruses comprise one of the members among 15 genera of the family Reoviridae (3). The genome of rotavirus consists of 11 double-stranded (ds) RNA segments contained within triple-layered, nonenveloped, icosahedral viral particles of about 100 nm diameter. Each RNA segment encodes one of the six structural proteins (VP1-VP4, VP6, and VP7) or six nonstructural proteins (NSP1-NSP6) (3,5). Based on the antigenic properties of the VP6, rotaviruses are classified into eight different serogroups from A–D and F–I (13). The outer protein layer is composed of the glycoprotein VP7 and the protease sensitive protein VP4, both containing epitopes for inducing neutralizing antibodies. These proteins also provide the basis for classification of rotaviruses into G and P types, respectively. The GARV 11 RNA segments comprise 220 genotypes, including 28 G genotypes and 39 P genotypes (13). The most prevalent GARVs detected in pigs are G3-G9 and G11 in combinations with either P[6] or P[7]. Furthermore, many VP7 types (G1, G2, G6, G8, G10, G12, and G26) and VP4 types (P[5], P[8], P[11], P[13], P[14], P[19], P[23], P[26], P[27], P[32], and P[34]) have also been detected sporadically in pigs (6,17).
Gene segment 6 encodes middle capsid protein VP6, which is highly conserved (∼87–92%) among mammalian GARVs (22). Therefore, these molecules are ideal for detections of GARVs through usual serological detection methods, including immunechromatographic assays, ELISA, immunofluorescence assay; as well as molecular diagnostic techniques, such as reverse transcription–polymerase chain reaction (RT-PCR) (7,15).
Due to the acute nature of rotavirus infection, a diagnostic technique that is less time-consuming, simple, sensitive, as well as accurate in detecting the causative agent is very important for the timely management of diseased animals to control further spread of infection to other animals (1). ELISA is commonly used for the detection of rotavirus due to their quicker nature of detection compared with other tests such as virus isolation (VI) (1) and RNA-PAGE (21). Some commercially available ELISA kits, primarily designed for human rotaviruses, have been reported to detect animal rotaviruses; however, they are not approved for use by veterinary diagnostic laboratories (1).
The objective of this study was to develop a sensitive and specific antigen-capture enzyme-linked immunosorbent assay (AC-ELISA) to detect rotavirus in porcine feces. In this work, we produced VP6 protein-based monoclonal (MAb) and polyclonal antibodies (PAb) and developed AC-ELISA for detection of porcine GARVs. This study will help in rapid diagnosis and timely management of porcine diarrhea caused by GARVs.
Materials and Methods
Ethics
The present study and the protocols used in the study were approved by the Committee on the Ethics of Animal Experiments at the College of Veterinary Medicine, Huazhong Agricultural University. The authors declare their compliance to publication ethics. This work does not include any studies performed on human participants.
Cells, viruses, plasmids, and bacteria
The rhesus monkey kidney epithelial cell line MA104 and murine Ag8 myeloma (SP2/0) cells were used. MA-104 cell monolayer was grown in 25-cm2 flasks (Corning, NY) in growth media comprising minimum essential medium (MEM; Gibco, Life Technologies), 10% fetal bovine serum (heat inactivated) (FBS), (Gibco, Invitrogen), 100 μg/mL streptomycin, and 100 IU/mL penicillin (Pen Strep; Gibco, Life Technologies) at 37°C with 5% CO2. SP2/0 cells were cultured in the RPMI-1640 medium (Gibco, Life Technologies), supplemented with 10% heat-inactivated FBS (Gibco, Life Technologies), 100 μg/mL streptomycin, and 100 IU/mL penicillin (Pen Strep; Gibco, Life Technologies) at 37°C with 5% CO2. Porcine rotavirus-A (PoRVA) TM-a strain (RVA/Pig-tc/CHN/TM-a/2011/G9P23), isolated in our laboratory in 2011, was used to prepare the template for cloning VP6 (GenBank: JF970185). Prokaryotic expression plasmid pGEX-KG (ATCC® 77103™) was used for cloning, and expression of VP6 protein in cloning and expression of Escherichia coli host strains DH5α and JM105, respectively. Porcine epidemic diarrhea virus (PEDV, CH/YNKM-8/2013, GenBank: KF761675.1), transmissible gastroenteritis virus (TGEV, WH-1, GenBank: HQ462571.1), pseudorabies virus (PRV, HNB, GenBank: KM189914), and porcine circovirus type 2 (PCV-2, Wuhan strain, GenBank: JF598044), available in our laboratory, were used for checking the specificity of our developed AC-ELISA test.
Cloning, expression, and purification of recombinant VP6
Porcine GARV strain TM-a (RVA/Pig-tc/CHN/TM-a/2011/G9P23) was propagated in 80% confluent monolayer of MA104 cells supplied with MEM containing 10 μg/mL trypsin (Gibco, Life Technologies), until obvious cytopathic effect (CPE) was observed. The infected MA104 cell monolayer was freeze-thawed three times at −80°C, to release the virus into the medium, and centrifuged at 10,000 g for 5 min (Eppendorf, Germany). About 200 μL of clarified supernatants were transferred into a separate sterile tube for total RNA extraction by using TRIzol reagent (Invitrogen), using the manufacturers' guidelines, followed by denaturing step at 95°C for 2 min, and instantly stored at −80°C till further use.
The porcine GARV-VP6 gene-specific forward (VP6-F1:5′-ATGGAGGTTCTGTACTCATT-3′) and reverse primers (VP6-R1:5′-CCCGTCGACTCACTTAATCAACATGCTTC-3′) were designed according to published sequence (DQ204741). RT was performed by using TaKaRa PrimeScript RT Master Mix (TaKaRa, Japan), containing PrimeScript RTase, RNase inhibitor, random hexamers, oligo dT primer, dNTP mixture, and reaction buffer, using the manufacturers' instructions. In brief, 10 μL reaction mixture was prepared by adding 8 μL template RNA and 2 μL 5 × PrimeScript buffer. Reverse-transcription reaction protocol was as follows: 37°C for 15 min, 85°C for 30 sec, and 4°C for 1 min. PCR was conducted to amplify a 1,194-nt long segment of VP6 from RT products using TaKaRa Ex Taq DNA polymerase (TaKaRa) according to the manufacturers' protocol. PCR was conducted in a 50 μL PCR reaction mixture tube (0.25 μL TaKaRa Ex Taq, 5 μL 10× Ex Taq buffer, 4 μL MgCl2 (25 mM), 4 μL dNTP mixture (2.5 mM each), 2 μL cDNA, 1 μL of each forward and reverse primers, and sterilized distilled water up to 50 μL) at 95°C for 5 min; total 35 cycles of 94°C for 30 sec; 55°C for 40 sec, 72°C for 1 min, and final extension at 72°C for 10 min.
The amplified product was gel-purified (Bioflux, Japan) and ligated into the prokaryotic expression vector pGEX-KG to produce pGEX-KG-VP6 construct, which was sent to GenScript Company (Nanjing, China) for sequencing. The recombinant vector plasmid pGEX-KG-VP6 and blank control vector plasmid pGEX-KG were separately transformed first in a competent cloning E. coli host DH5α and then in a competent expression host JM105. The transformants were cultured in Luria–Bertani (LB) broth supplemented with ampicillin (50 μg/mL) in a shaking incubator (180 g) at 37°C for about 3 h. When the OD600 value reached 0.5–0.6, isopropylβ-D-1-thiogalactopyranoside (IPTG) was added to the medium to give a final concentration of 1 mM to induce protein expression. They were further cultured for 4 h, after which the cultures were centrifuged at 8,000 g, at 4°C for 5 min. The bacterial pellets were resuspended and sonicated to obtain a clear lysate. The expressed recombinant VP6 (rVP6) and blank control plasmid suspensions were separately purified using Glutathione Sepharose 4B affinity chromatography column (GE Healthcare, Sweden) as per manufacturers' instructions and stored at −80°C.
Polyclonal antibody production
Ten-week-old New Zealand white rabbits were used for PAb production. About 1 mg (1 mg/mL) purified, rVP6 was emulsified in an equal volume of Freund's complete adjuvant (FCA), and injected subcutaneously at multiple sites. Booster doses were administered at 2-week intervals with 1 mg of the same antigen emulsified in Freund's incomplete adjuvant (FIA). Ten days after the third booster dose, the serum samples were collected and checked for antibody titers by indirect ELISA, using rVP6 (1 μg/well) as coating antigen. PAb were purified using commercial protein-A affinity purification kit (GE, Sweden) using the manufacturers' guidelines.
Monoclonal antibody production and purification
Four-week-old BALB/c mice (n = 5) were immunized with 100 μg of rVP6 emulsified in FCA. Three booster doses were administered with the same amount of rVP6 emulsified in FIA, at 2-week intervals. The same dose was injected intraperitoneally 3 days before cell fusion experiment. Serum samples were collected and checked for titers for antibody against rVP6. Mice with higher titers were selected for cell-fusion experiment. The spleens were dissected to obtain the splenocytes, which were mixed with Sp2/0 myeloma cells (at a ratio of 5:1) in 1 mL 50% (w/v) polyethylene glycol (MW 1500; Roche, Mannheim, Germany) for 1 min at 37°C. They were serially diluted in RPMI-1640, containing 20% FBS (Gibco, Life Technologies), and hypoxanthine–aminopterin–thymidine medium (HAT; Sigma, MO) and plated in 96-well plates (Corning, NY).
Indirect-ELISA, western blot analysis, and indirect fluorescence assay were performed according to the methods reported previously (14) to screen for positive hybridomas against rVP6 protein and porcine rotavirus-A, which were cloned thrice by limiting dilution method. Only those hybridomas, which exhibited strong positive signals against both rVP6 and PoRVA and negative signals against purified proteins obtained from blank control plasmid, were further selected for each subcloning step. MAbs were isotyped using Pierce Rapid ELISA Mouse mAb Isotyping Kit (Thermo Fisher Scientific) using the manufacturers' protocol. Superior hybridoma clones were injected intraperitoneally into paraffin-primed BALB/c mice. Ten days later, ascites were harvested and IgGs were precipitated in saturated ammonium sulfate, followed by dialysis and purification on a Protein-G affinity column (GE, Sweden) using the manufacturers' instructions.
AC-ELISA development
A checkerboard titration was carried out to select the optimal concentrations of rabbit anti-rVP6 PAb (capture Ab), mice anti-VP6-MAb (detection Ab), and HRP-goat anti-mouse MAb by using rVP6 as a positive control. All 96 wells of the polystyrene microtiter ELISA plate were coated with 250 ng of rVP6 rabbit PAb in 100 μL coating buffer (0.05 M sodium carbonate–bicarbonate buffer, pH 9.6). Blocking was done with 200 μL of blocking buffer (3% BSA diluted in phosphate buffer saline +0.05% Tween-20; PBST) to cover free sites and prevent nonspecific binding. The plates were incubated with 50 μL/well of a 10% (w/v) fecal suspension, 100 μL mouse anti-VP6 MAb (100 ng/well) diluted in blocking buffer (1% BSA), HRP goat anti-mouse IgG 1: 10,000 (ABclonal) 100 μL/well (diluted in blocking buffer, 1% BSA). Each step received incubation at 37°C for 1 h, followed by washing four times with wash buffer (PBST). Finally, 100 μL tetramethylbenzidine was added to each well for color development. Absorbance was measured using ELx800 Absorbance Reader (BioTek, Winooski, VT) at a wavelength of 630 nm. The cutoff value was set at mean absorbance value of 25 porcine GARV negative fecal samples plus 3 × standard deviations (SD).
Analytical specificity and sensitivity of the AC-ELISA
Diarrhea-causing as well as other common porcine viruses, such as PEDV, TGEV, PRV, PCV2, were used to check the specificity of AC-ELISA. Porcine GARV TM-a strain was used as positive control and PBS was used as negative control.
For determining the analytical sensitivity, GARV-infected cell cultures (106.5TCID50/mL) were serially diluted 10-fold in MEM. Each dilution mixture (100 μL) was evaluated by AC-ELISA. The absorbance value just above the cutoff value was considered the detection endpoint of the AC-ELISA. The serial 10-fold dilutions were also subjected to RT-PCR by using GARV-specific forward primer GARV-F 5′-AAAGATGCTAGGGACAAAATTG-3′ and reverse primer GARV-R 5′-TTCAGATTGTGGAGCTATTCCA-3′ targeting a 309-bp-conserved GARV VP-6 gene segment according to a previously reported method (20).
Validation of AC-ELISA
From December 2015 to February 2016, 295 fecal samples of piglets exhibiting clinical diarrhea symptoms were collected from 17 farms in the Hebei, Henan, Hunan, Beijing, and Hubei provinces of China. The samples were transported in sterile plastic cups on ice to the Diagnostic Laboratory for Animal Diseases, Huazhong Agricultural University, Wuhan, China for evaluation and validation of the AC-ELISA. The samples were diluted in PBS to make 10% (w/v) fecal suspensions, vortexed for 10 min, and centrifuged at 10,000 g (Eppendorf, Germany) for 10 min; supernatants were subjected to AC-ELISA, conventional RT-PCR, and our in-house TaqMan fluorescence RT-quantitative PCR (qPCR).
RT-qPCR reaction was carried out in Applied Biosystems 7300 qPCR system (Applied Biosystems, Thermo Fisher Scientific, Foster City, CA), using THUNDERBIRD® Probe qPCR Master Mix kit (Toyobo, Osaka, Japan). RT-qPCR assay was performed in a reaction system of 20 μL containing 2 μL of cDNA, 10 μL Probe qPCR Mix, 300 nM of each F2 (VP6-F2: 5′-TTTATAGATAATGTATGTATGGATG-3′) and R2 (VP6-R2: 5′-CCAATTCTCAATGTAATCAG-3′) primers, 150 nm probe (5′-AATAGCCAGAATCAACGAA-3′), 200 nM 50× ROX reference dye, in 96-well plate, at 50°C for 2 min, 95°C for l0 min, 95°C for 15 sec, 52°C for 30 sec, and 72°C for 31 sec, 40 cycles, where each template was repeated three times, then the average result was selected for analysis. The results were subsequently compared for relative sensitivity, specificity, and coincidence value.
Results
Cloning, expression, and purification of rVP6
VP6 (1,194 bp) was cloned from porcine GARV TM-a strain (Fig. 1A) and ligated in prokaryotic expression vector to form pGEX-KG-VP6 (Fig. 1B). After expression and purification, the resultant rVP6 was obtained of ∼70 kDa (Fig. 1C).
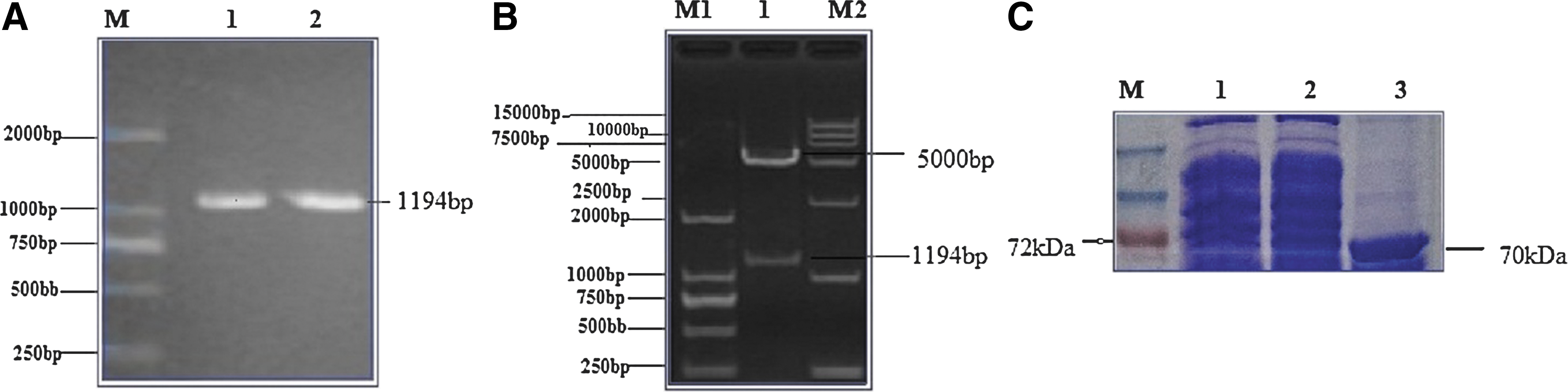
Cloning, ligation, and expression of rVP6.
Polyclonal and monoclonal antibody production and purification
PAb and MAb were successfully produced against VP6 of PoRVA TM-a strain. A panel of two superior hybridoma clones secreting MAb 1A7 (IgG1) and 3F5 (IgG2a) were selected on the basis of their strong reaction with both VP6 and PoRVA in indirect-ELISA. However, antibody titer of 3F5 was twofold higher (1:25,600) than that of 1A7 (1:12,800); therefore, it was selected as the detector antibody for AC-ELISA. PAb also had higher antibody titers (1:25,600) and reacted well against PoRVA. The reactivities of the both MAb and PAb were further verified by western blot analysis and indirect fluorescence assay (Fig. 2).
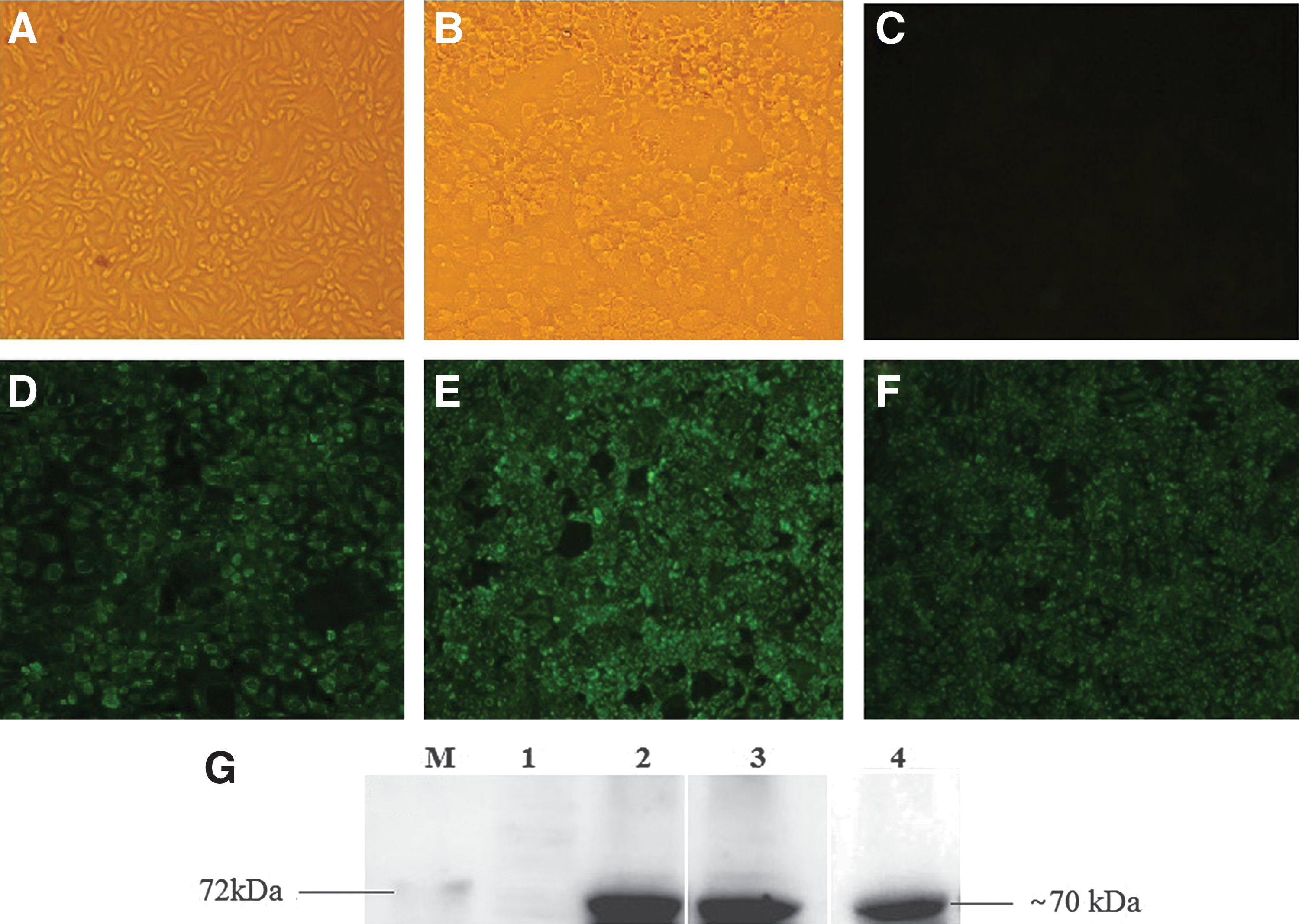
Reactivity of PAb and MAb.
AC-ELISA development
A checkerboard titration was undertaken to select optimal concentrations of different combinations of antibodies and reagents. It was found that 250 ng/well rabbit anti-rVP6 PAb was optimal as coating antibody; 100 ng/well 3F5 mice anti-VP6-MAb was optimal as detection Ab, and 1:10,000 dilution of the HRP-goat anti-mouse MAb was found to be optimal for the test. The cutoff value was calculated as mean absorbance of 25 porcine GARV negative fecal samples plus 3 × SD, and was set at 0.45 OD 630 nm.
Analytical specificity and sensitivity of the AC-ELISA
Analytical specificity of our developed AC-ELISA was evaluated using diarrhea-causing and other common porcine viruses (Fig. 3). PoRV, used as positive control, exhibited a strong positive reaction, while other porcine viruses and the negative control failed to exhibit a positive reaction. To check the detection limit, analytical sensitivity of AC-ELISA was determined by applying serial 10-fold dilutions of the GARV-infected cell cultures (106.5TCID50/mL) and compared with that of RT-PCR. The analytical sensitivity of AC-ELISA was found equal to that of RT-PCR, about 102.5 TCID50/mL (Fig. 4).
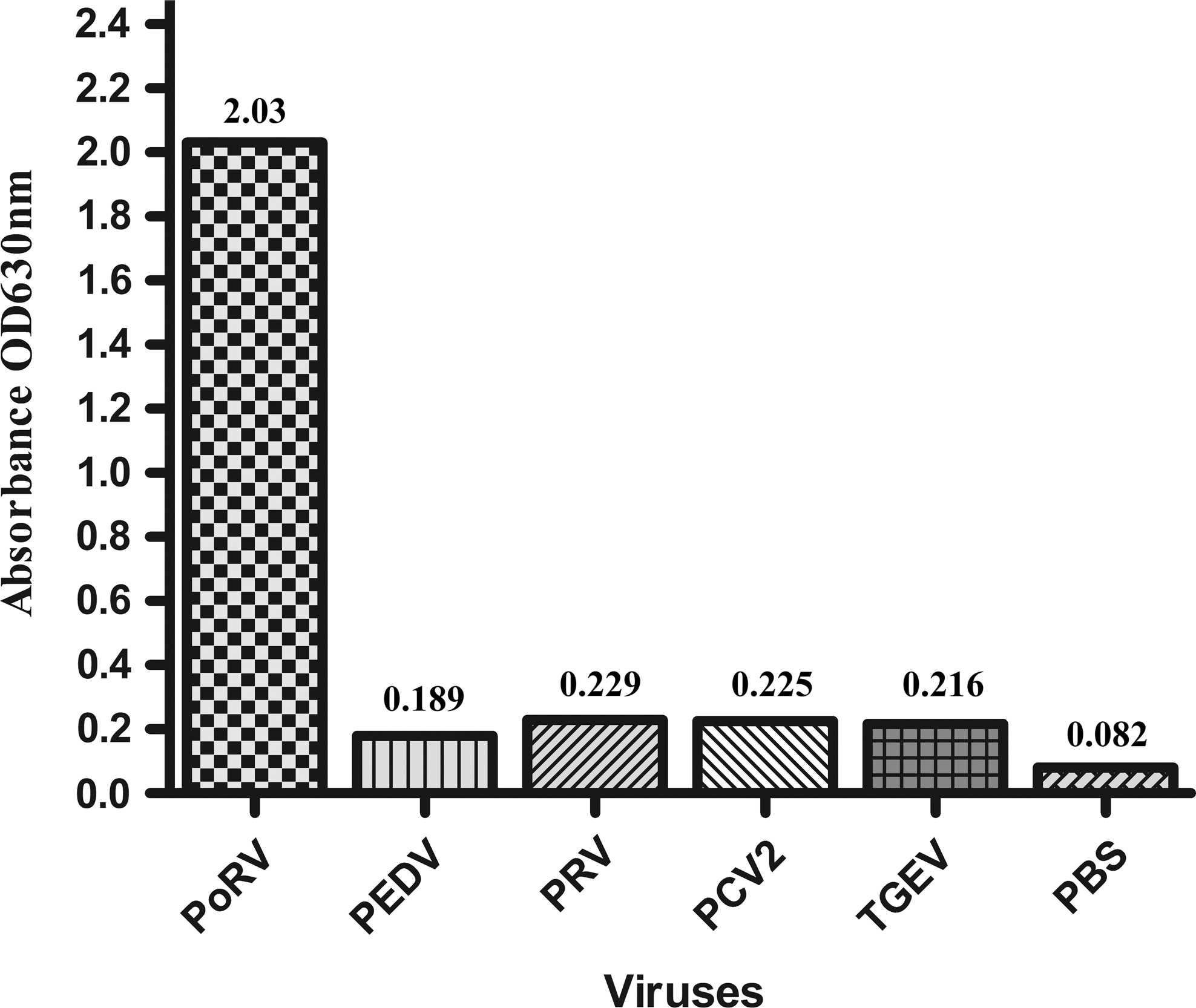
Analytical specificity of AC-ELISA. AC-ELISA, antigen-capture enzyme-linked immunosorbent assay.
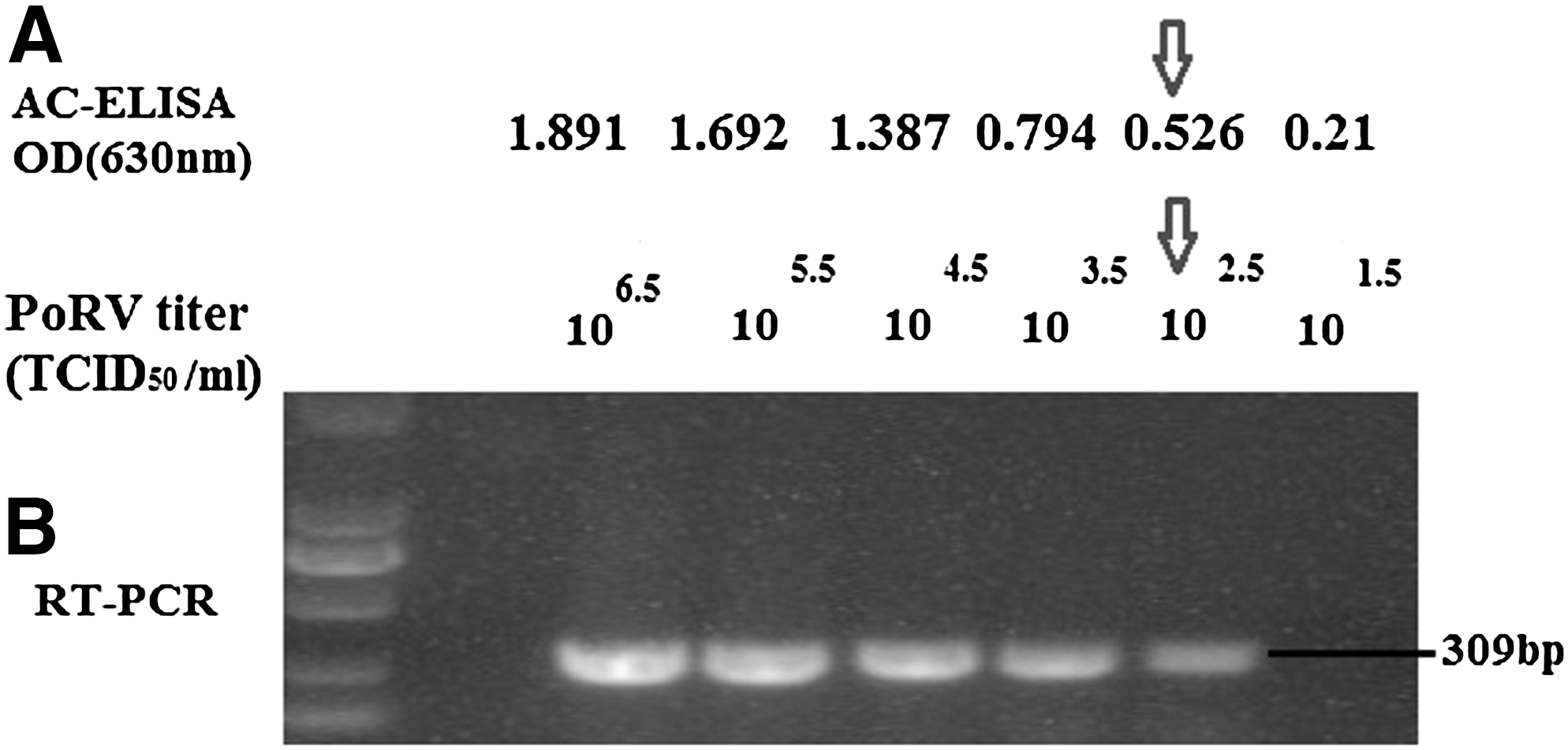
Analytical sensitivity of AC-ELISA compared with that of RT-PCR 10-fold serial dilution of GARV-infected cell cultures was subjected to both AC-ELISA
Validation of AC-ELISA
Total 295 porcine fecal/diarrhea samples received from the field were subjected to AC-ELISA, conventional RT-PCR, and TaqMan RT-qPCR. The results revealed that 11.2%, 11.5%, and 12.2% samples were detected as PoRVA positive with AC-ELISA, conventional RT-PCR, and TaqMan RT-qPCR, respectively (Supplementary Table S1; Supplementary materials are available online at
The relative sensitivity and specificity of AC-ELISA, compared with those of RT-PCR were found to be ∼95.8% (95% confidence interval: 0.8467–0.9993) and 100% (95% confidence interval: 0.9860–1.000), respectively. The coincidence coefficient value between AC-ELISA and RT-PCR was ∼99.6 (95% confidence interval: 0.978–0.999), with a kappa (κ) value of 0.972, which indicates strong agreement between these two techniques (Table 1). Whereas, the relative sensitivity and specificity of AC-ELISA, compared with those of TaqMan RT-qPCR, were found to be ∼91.7 (95% confidence interval: 0.775–0.983) and 100% (95% confidence interval: 0.986–1.00), respectively. The total coincidence value between AC-ELISA and TaqMan RT-qPCR was ∼98.83 (95% confidence interval: 0.967–0.998), with κ value of 0.95, which indicates strong agreement between these two techniques (Table 1).
Comparison of AC-ELISA with RT-PCR: The relative sensitivity is 33/(33 + 1) × 100 = 97.059%; relative specificity: 261/(0 + 261) × 100 = 100%; concordance: (33 + 261)/295 × 100 = 99.66%; the kappa: (Po-Pe)/(1-Pe), where, Po = (33 + 261)/295 = 0.996; Pe = (34/295) × (33/295)+(261/295) × (262/295) = 0.799, kappa = 0.996–0.799/1–0.799 = 0.98.
Comparison of AC-ELISA with TaqMan qRT-PCR: The relative sensitivity is 33/(33 + 3) × 100 = 91.667%; relative specificity: 259/(0 + 259) × 100 = 100%; concordance: (33 + 259)/295 × 100 = 98.983%; the kappa: (Po-Pe)/(1-Pe), where, Po = (33 + 259)/295 = 0.989; Pe = (36/295) × (33/295) + (259/295) × (262/295) = 0.793, kappa = 0.989–0.793/1–0.793 = 0.95.
AC-ELISA, antigen-capture enzyme-linked immunosorbent assay; RT-PCR, reverse transcription–polymerase chain reaction; TaqMan RT-qPCR, TaqMan fluorescence quantitative reverse transcription–polymerase chain reaction.
Discussion
Diarrhea is considered to be a very common health problem, leading to high economic losses to the porcine industry (11). PoRVA is one of the common causes of mild to severe dehydrating diarrhea in both the enzootic and epizootic forms, leading to losses in weaning and postweaning piglets. PoRVA is widely reported in China, secondary to PEDV, as a causative agent of viral diarrhea, but is still a considerable threat to the Chinese porcine industry (15,19,26). Recently, many published studies from China have evidenced transmission between porcine and human rotavirus (15,29). With the passage of time, RVA evolves in their molecular epidemiology, which causes failure in the efficacy of routinely used field vaccines. Therefore, continuous surveillance of PoRVA is very important in China, to ensure awareness of the different circulating strains, groups, and serotypes of rotavirus reservoirs that may lead to the emergence of new rotavirus strains or genetic elements in humans or other animal populations.
Rapid, accurate, cost-effective, and less laborious diagnostic kits are very helpful in surveillance, timely management, and control of acute infections such as rotavirus diarrhea. Currently, there is no any gold standard detection method for detection of PoRVA. For example, diagnostic methods with high specificity, such as VI, immunohistochemistry and electron microscopy are time-consuming, require skilled technical staff, and VI has risk of contamination. Molecular methods for detecting the viral genome, such as RT-PCR and real time PCR are highly sensitive, but such techniques are not cost-effective and cannot be practically used in the field (8). However, the AC-ELISA is swifter and highly capable of large-scale screening of field samples, and has been reported to provide benefits over other traditionally used antigen capture techniques (10).
Specificity of AC-ELISA is generally improved by use of MAb (2). However, PAbs are preferred by many researchers to increase sensitivity of the assay (4,14). In the current study, we have used a wild-type PoRVA strain (RVA/Pig-tc/CHN/TM-a/2011/G9P23), which was isolated from a piglet exhibiting severe diarrhea, dehydration, and vomiting symptoms, in Wuhan, China, in 2011. Furthermore, sequencing results reveal that VP6 of this Chinese strain (RVA/Pig-tc/CHN/TM-a/2011/G9P23) belongs to I5-genotype, which is most dominant in pigs, particularly in China. Due to these properties, this antigen is an ideal choice for development of the AC-ELISA for detection of PoRVA.
In our study, we developed an AC-ELISA for detection of PoRVA. Both rabbit PAbs and murine 3F5 MAbs were produced against PoRVA-VP6. We tried different MAbs and PAbs combinations for optimization of AC-ELISA, including murine 3F5 MAbs as capture Ab and rabbit PAbs as detector Ab; and rabbit PAbs as capture Ab and murine 3F5 MAbs as detector Ab. Both combinations were highly specific in detecting PoRVA. However, later one was found to be more sensitive, therefore, finally selected for this AC-ELISA. Moreover, there is no earlier published report of the use of both MAbs and PAbs produced against VP6 for developing an AC-ELISA against PoRVA. In our study, rVP6 was preferred over whole PoRVA for the production of PAbs, because of the large variations in surface proteins (VP4 and VP7) among different PoRVA strains, which may be an important factor in enhancing both sensitivity and specificity of our developed method. A similar phenomenon has also been reported for influenza viruses (9).
For validation of AC-ELISA, 295 porcine fecal samples were used for detection of PoRVA. Totally, 33 samples were found positive by AC-ELISA. However, three more samples were found positive by TaqMan RT-qPCR. Failure in detection of these three samples may be linked to some nonspecific factors present in diarrhea samples that may block the binding of antigen with capture antibody or relatively lower sensitivity of AC-ELISA than RT-qPCR. The analytical sensitivity of AC-ELISA was found to be equal to that of RT-PCR, about 102.5 TCID50/mL. Moreover, AC-ELISA failed to detect any cross-reactivity with PEDV, TGEV, PRV, and PCV2 viruses.
Different epidemiological studies have shown that prevalence of PoRVA is highly variable throughout the world, which may be due to the number of clinical samples used in these studies, the age and health of animals, season, and geographical distribution. Theuns et al. (24) found the prevalence of PoRVA was as high as 49% in Belgium. Papp et al. (17) reviewed the prevalence of PoRVA across the world and summarized results revealed that 24% of clinical samples were detected as positive. Song et al. (20) explored that PoRVA was prevalent in 13.2% collected samples in South Korea. Zhou et al. (28) reported the prevalence of PoRVA is 0.9% in five European countries, including Austria, Germany, Hungary, Sweden, and Spain. Unfortunately, a few epidemiological studies have been conducted to check prevalence of PoRVA in China. However, the prevalence of PoRVA reported in recent two extensive epidemiological studies conducted by Zhang et al. (27) and Zhang et al. (26) are much lower, about 9.6% and 11.6%, respectively. In our study, the total detection rate of PoRVA was 11.2% with AC-ELISA, which is similar to these two recent epidemiological studies from China.
Conclusion
In conclusion, we successfully developed swift, highly specific, and sensitive VP6 MAb-based AC-ELISA that efficiently detected PoRVA in fecal samples. This assay will be effective in the timely detection and surveillance of rotavirus to initiate proper preventive measures for disease management.
Footnotes
Acknowledgments
This work was supported by the Fundamental Research Funds for the Central Universities (No. 2662015PY141) and China Agricultural Research System (No. CARS-36).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.