
Abstract
Host hepatitis C virus (HCV)-specific T cell responses and the ability of the virus to escape this response are important correlates of infection outcome. Understanding this host–viral interplay has been difficult given the often asymptomatic nature of acute HCV infection. We studied a recent transmission case to determine whether adapted viral strains can be transmitted and influence the recipient's anti-HCV T cell response. The diversity of viral populations was examined using next-generation sequencing, and HCV-specific T cell interferon (IFN)-γ responses were assessed using a peptide panel representing the autologous viruses. HCV-specific T cell responses in the source were directed against peptides that did not match the dominant autologous virus but rather low-frequency variants, implying existing viral adaptation in the source strain. Most HCV T cell epitopes that elicited an IFN-γ response in the source did not in the recipient, despite the pair sharing human leukocyte antigen alleles that govern antigen presentation and similar autologous viruses. Intrahost HCV variation in the recipient fell within predicted T cell epitopes, suggesting alternative targets of the immune response. These data suggest that transmission of adapted viral species can direct the host's HCV-specific immune response profile during acute infection.
Introduction
H
Rapidly mutating viruses such as HCV evolve within a single host to increase replicative fitness and escape host immune pressure with an estimate of replacement of the original variant up to 0.1 per day (3). This rapid evolution of HCV following transmission is followed by a reduction in diversity suggestive of a second genetic bottleneck at ∼100 days postinfection, also likely driven by host immune pressure (4). At a population level, HCV evolution is also shaped by the sequential infection of different hosts within a population (12,25); this form of adaptation is observed at the genetic level as HLA footprints (16). These HLA footprints are identified via the association of specific amino acid variations within the HCV genome and carriage of particular HLA alleles. These HLA-associated viral polymorphisms fall within or flank known HCV T cell epitopes and accordingly reflect in vivo T cell pressure (12,23,25).
Despite this overall evolution of HCV within a host population, previous work from our group indicated that acute HCV infection is characterized by fixed mutations with a bias for silent changes, likely reflecting structural and functional constraints within the virus that may limit its ability to escape T cell responses relative to a more mutable virus such as HIV (8,24). Such a pattern of evolution for HCV may result in the transmission and maintenance of viral immune escape mutations in a new host, particularly in the presence of compensatory mutations that offset fitness (10,20,22,27). The transmission of viral adaptations is especially relevant when both the source and the recipient share part of their HLA repertoire and, in such cases, it would be expected that the transmission of viral species carrying immune escape mutations may affect the selection of viral targets for the HCV-specific T cell immune response in the new host during the critical acute stage of infection.
Understanding the dynamics of the host–viral interplay in the setting of a recent transmission has been difficult due to the often-asymptomatic nature of acute HCV infection. As the source of the transmitted virus is typically unknown, assumptions on the characteristics of the original infecting virus have been made in acute HCV infection studies (4,34). In this study, we studied a couple known to have shared needles exclusively between themselves and during the acute phase of infection of the previously HCV seronegative recipient. The couple also shared a subset of their HLA allele repertoire. Next-generation sequencing (NGS) and immunological analysis of samples during the acute phase of infection in the recipient and the time point close to transmission for the source allowed an analysis of the impact of existing adaptation in the transmitted viral population to subsequent immune target selection in the new host.
Materials and Methods
Ethics statement
Written, informed consent from subjects was obtained for this study. Ethics approval for the conduct of this research was obtained from the Royal Perth Hospital (Perth, Australia) Ethics Committee (EC2004/005). The protocol and the procedures of the study were conducted in conformity with the ethical guidelines of the World Medical Association Declaration of Helsinki.
Subjects
The transmission pair studied consisted of two injecting drug users (IDUs). The source was infected with an HCV genotype 1a strain before the presentation of the recipient and stated the couple shared needles with each other exclusively. Phylogenetic analysis of HCV sequences from the pair and HCV-infected subjects from the same geographical location, including IDUs, confirmed a recent transmission event (Supplementary Data; Supplementary Data are available online at
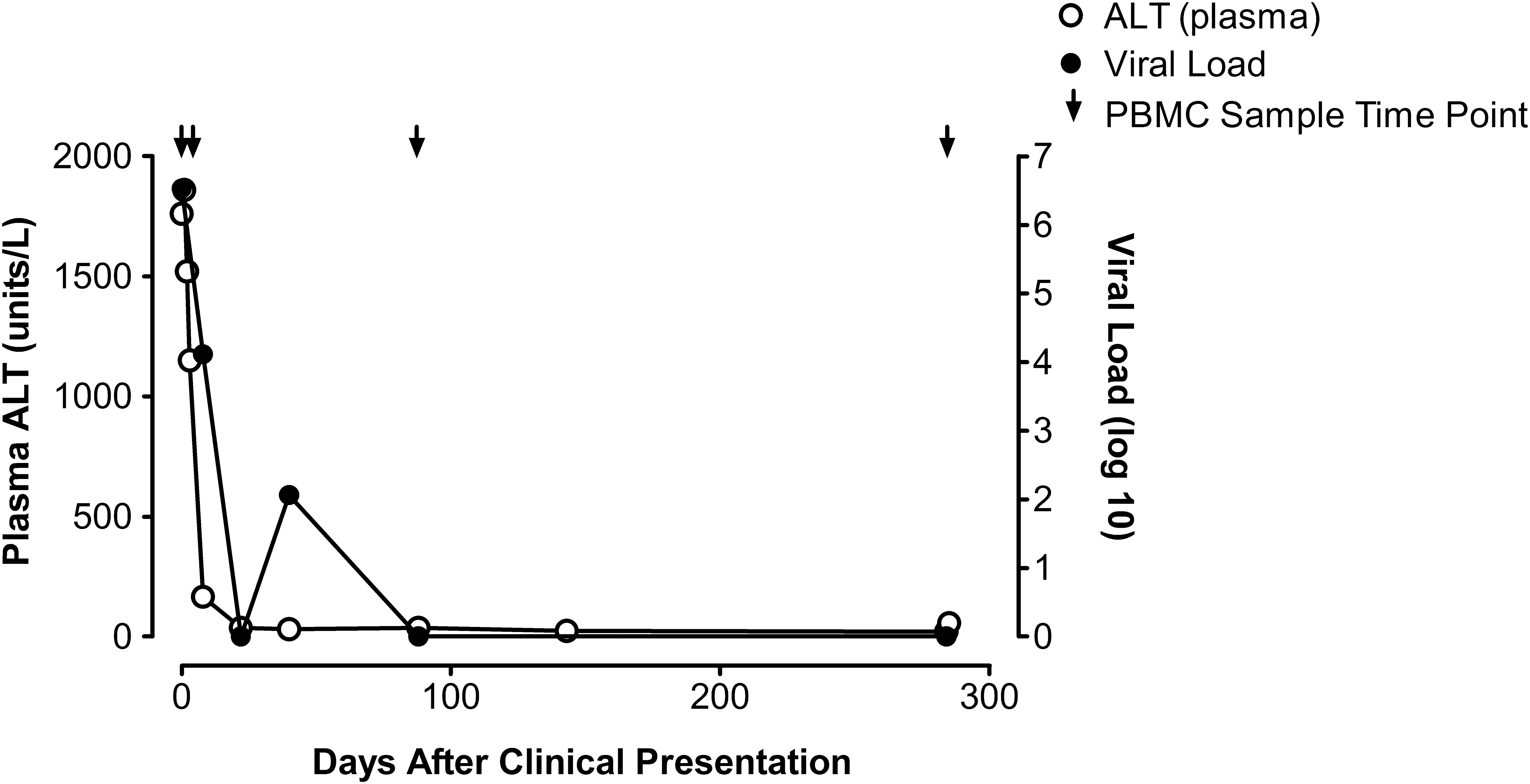
High plasma ALT and HCV viral load in the recipient suggest sampling during acute HCV infection. Viral load decreased over time to below detectable levels reflecting viral clearance. Samples obtained at day 3 after initial clinical presentation of the recipient were used for bulk and NGS. Undetectable viral load was found at days 8, 88, and 284 postclinical presentation. Detection limit for the viral load assay was 15 IU/mL. ALT, alanine aminotransferase; HCV, hepatitis C virus; NGS, next-generation sequencing.
Host genotyping
HLA typing for HLA-A and -B was performed using sequence-based methods as previously described (12). Typing for the interferon lambda 4 (IFNL4) rs12979860 single-nucleotide polymorphism (SNP) was performed using a TaqMan probe real-time method as described elsewhere (31). See Table 1 for host genotyping results.
Subjects share underlined HLA alleles.
HLA, human leukocyte antigen; IFNL4, interferon lambda 4.
HCV sequencing
Viral RNA used for sequencing was extracted from plasma samples using the COBAS AMPLICOR HCV Specimen Preparation Kit v2.0 (Roche, Sydney, Australia) according to the manufacturer's instructions.
Polymerase chain reaction (PCR) amplification and Sanger-based sequencing of the HCV nonstructural proteins were performed as previously described (25). Mixtures were identified where the secondary peak was ≥20% of the major peak. Due to the variability of the HCV genome, some samples failed to produce a PCR product for some region(s). The HCV sequences in this study have been submitted to GenBank with the following accession numbers: JQ417892–JQ417901.
NGS of the HCV NS5B region was carried out using the 454 Life Sciences platform (GS-FLX; Roche Applied Science, Penzburg, Germany) for both subjects, using PCR amplicons from above according to the manufacturer's instructions. Nucleotide data were analyzed using the Genome Sequencer software (Roche Applied Science) and in-house programs to evaluate the quality of sequence reads and correct for likely errors in homopolymer stretches. All sequence reads were aligned to the H77 genotype 1a sequence (GenBank accession No. NC004102). Source and recipient median NGS read coverage for NS5B was 3,022 (interquartile range [IQR] 2,556–8,856) and 1,754 (IQR 1,164–2,200), respectively. Only variants detected with a frequency of >1% were considered for analyses.
For both the source and recipient, viral sequences were obtained from samples collected at day 3 after initial clinical presentation of the recipient.
Peptides used in IFN-γ enzyme-linked immunospot assay
IFN-γ enzyme-linked immunospot (ELISPOT) assays were performed on PBMCs from the transmission pair using peptides representing T cell epitopes for the HLA class I alleles of the subjects. The panels of known HCV T cell epitopes used in this study were all within the NS2-NS5B region and included HCV T cell epitopes that had been predicted from statistically significant HLA allele-associated HCV polymorphisms identified by a population-based genetic analysis of NS2-NS5B sequences from chronic HCV genotype 1a-infected individuals (including from the same geographical region as the transmission pair) (23) and from the Immune Epitope Database (IEDB) (33). The peptide sequences for the T cell epitopes were based on consensus sequence (identified in the population-based study) and variants (most common sequence variants identified in the cohort) identified from the large genetic-based study (25) and included peptides corresponding to the autologous sequence in the subject and recipient (see list of tested peptides in Table 2).
Unless indicated, T cell epitope sequence from Pfafferott et al. (23).
T cell epitope listed in IEDB. For position NS5B 2466, T cell epitope listed in Ruhl et al. (28) (genotype 1B).
+, Indicates matching sequence; IEDB, Immune Epitope Database; N/A, sequence not available.
IFN-γ ELISPOT assay
IFN-γ ELISPOT assays were performed using the Biomek FX (Beckman Coulter, Indianapolis, IN) liquid handling workstations as previously described (1) with the modification of the addition of 200,000 PBMCs per well. Wells containing PBMCs and anti-CD3 antibodies (Mabtech, Nacka, Sweden) were used as positive controls, while negative controls consisted of PBMCs cultured in media alone (triplicate). IFN-γ producing spots were enumerated using an AID ELISPOT reader (Autoimmun-Diagnostika, Strassberg, Germany). The number of IFN-γ producing T cells was determined after subtraction of the background (defined as the mean plus three times the standard deviation of the number of spots counted in the negative control wells).
Peptide prediction for sites of viral variation between source and recipient
The sequences of regions containing a variant between the source and recipient of the transmission pair were entered into NetMHC (21) to identify putative CD8+ T cell epitopes (suggested binding peptide indicated when score below 500 nM IC50).
Results
HCV sequences from subjects support a recent transmission event and subsequent bottleneck in the recipient
Phylogenetic analysis of the Sanger-based sequence from the two subjects reveals a high level of sequence identity and close phylogenetic relatedness relative to HCV sequences from HCV-infected subjects in the same geographical location supporting a recent transmission event (Supplementary Data and Supplementary Fig. S1).
To further characterize the dynamics of the transmission event, the diversity of the viral populations in the subjects was examined using NGS data for the NS5B region as previously described (4) (Supplementary Data). The analysis identified 15 and 10 viral variants (haplotypes) for the source and recipient, respectively (at a frequency of >1%) (Supplementary Fig. S2). Phylogenetic comparison of these NGS haplotypes showed a more diverse viral population in the source when compared to the recipient (Supplementary Figures S3 and S4), with a mean genetic diversity in the source and recipient haplotypes of 0.010 and 0.005, respectively (Shannon entropy was also greater between the source NGS haplotypes than for the recipient NGS haplotypes). Furthermore, a cluster of source viral strains were not represented in the recipient. Overall, these results support a transmission bottleneck and within host diversification in the recipient.
Identification of T cell responses in the source in regions of HCV known to contain viral adaptations
The HCV-specific T cell immune response profile of the transmission pair was studied using peptides covering T cell epitopes in the HCV NS2-NS5B region restricted by the HLA alleles carried by each individual and thus represents likely immune targets (Table 2). The source was only tested at a single time point during the chronic phase of HCV infection, but the recipient was tested at multiple time points during acute HCV infection (days 3, 8, 88, and 284 postclinical presentation). As the subjects both carried the common HLA class I alleles HLA-A*02:01 and HLA-B*44:02, most peptides tested were the same for the two subjects (39/43 peptides covering four HLA-B*44 and 12 HLA-A*02 T cell epitopes). Importantly, for the majority of T cell epitopes, the dominant autologous virus within each subject of the transmission pair was represented by a peptide in the screen (14/17 for the source and 12/15 for the recipient; Table 2).
All T cell responses detected in the subjects were directed against HLA-A*02-restricted T cell epitopes. Five IFN-γ T cell responses were detected in the source and only one response was identified in the recipient (Table 3). Both subjects showed T cell responses to the known HLA-A*02-restricted T cell epitope at NS31073–1081. However, the peptide matching the dominant autologous viral sequence (CINGVCWTV) in both subjects did not elicit a detectable response. In the recipient, this T cell response was detected at day 8 postclinical presentation and was maintained at a similar level up to day 284 (final time point tested) and after spontaneous clearance of the virus. However, there were insufficient PBMCs at the later time points for the recipient to test both peptides for this T cell epitope and we cannot exclude responses in the recipient in the later phase of acute infection. No additional T cell responses were observed in the recipient for T cell epitopes restricted by the shared HLA class I alleles HLA-A*02:01 and HLA-B*44:02 and tested in this study. Furthermore, the recipient did not show a response to the two T cell epitopes tested in the panel that were restricted by HLA-B*15:01; an HLA allele present only in the recipient (Table 2).
Variant amino acid between peptides in bold.
Epitope in IEDB or as otherwise noted.
Based on individual sequence reads that span epitope.
Days postclinical presentation.
Sequence based on sample obtained from day 3 (postclinical presentation).
Peptide not present in IEDB.
From Pfafferott et al. (23), P at position 4 in consensus is predicted adapted amino acid.
The source does respond to a form of the NS3 1406–1415 T cell epitope (KLVALGPNAV) that is not in the published data sets and is a form that is highly unlikely to have infected the source and is therefore not included in this analysis. The recipient does not show a response against this peptide.
NGS, next-generation sequencing; SFU, spot forming unit.
Of the five T cell responses detected in the source, two responses were for the same HCV T cell epitope in NS5B2727–2735 resulting in four distinct regions within HCV NS2-NS5B targeted by the source. As described for the HLA-A*02-restricted NS3 T cell epitope above, the peptide eliciting the response in three of these four regions did not represent the dominant strain in the contemporaneous plasma sample (Table 3) even though the peptide representing the autologous sequence was tested in the IFN-γ ELISPOT assay. In all cases, the recipient shared the same dominant amino acid as the source, with no observable T cell response to any peptides tested.
We then used NGS to characterize the viral quasispecies in the subjects to assess whether the observed disparity between the T cell responses and autologous viruses in the source was consistent with viral adaptation and/or the presence of low-frequency strains that may contain the sequence of the responding peptides. The frequencies of the strains containing the peptide sequences that elicited responses in the source were not present at levels above the cutoff of 1% (if detected then <0.1% and on only a few sequence reads) except for the peptide sequence DLPLIIQRL at the T cell epitope NS5B2878–2886 that was present at ∼2% in the source (Table 3). For the NS5B2878–2886 T cell epitope, the dominant strain in both the source and recipient contains the likely adapted form (DLPPIIQRL) of the epitope as determined by our population-based genetic study (23,25). For the other peptides eliciting a response based on known T cell epitopes for which we did not identify a putative adaptation site based on our earlier study, it is not always clear what peptide is the adapted form. Collectively, these results suggest that the dominant source sequence is adapted within several HLA-A*02-restricted T cell targets and these putative adaptations are present in the recipient as the dominant sequence despite a bottleneck during the transmission event.
Intrahost diversification following transmission event: identification of putative new immune targets in the recipient
Given that the transmission of adapted viruses in the transmission pair may have affected the recipient's HCV-specific T cell responses, we sought to determine whether sites of amino acid difference between the source and recipient sequence (as determined by Sanger-based sequencing) fell within predicted or known T cell epitopes, representing de novo adaptation to other T cell targets in the recipient. Identification of such sites would suggest signatures of strong in vivo T cell immune pressure or reversion of viral adaptations as only variants at >20% of the dominant strains were identified (reflecting rapid fixation or major shift in variant frequency within the recipient by day 3 postclinical presentation or likely within 6 weeks from infection). There were 15 amino acid sites differing between the source and recipient within the HCV NS2-NS5B region. Of these sites, 1 fell within a known T cell epitope with the relevant HLA restriction and 12 other sites fell within predicted T cell epitopes (Table 4).
HLA-A*30 indicates HLA-A*30:01 in the NetMHC v3.4 program not HLA-A*30:04 (present in source). HLA-B*14:02 was used in NetMHC as HLA-B*14:01 was not available for peptide binding affinity predictions.
Source (S) and recipient (R). Binding score in nM and >500 nM IC50 suggestive of a nonbinding peptide, >50 and <500 nM suggestive of weak binder, and <50 nM suggestive of strong binder.
T cell epitope from Pfafferott et al. (23) and in peptide screen.
Many of the differences between the pair were due to the presence of two amino acids (mixture) at the same position within the source sequence but a single amino acid in the recipient. At some of these sites, the amino acid within the recipient virus had a predicted lower binding affinity for a peptide restricted by a HLA allele shared by the pair (Table 4). These results may reflect continued immune pressure on matched HLA-restricted T cell epitopes or new T cell targets for the recipient. T cell immune responses against these regions were not tested (aside from the single known T cell epitope) as these peptides were not included in the original screen for HCV-specific T cell responses and there were limited PBMC samples for the subjects available for further testing.
Some of the differences between the dominant sequences of the pair fell within a predicted T cell epitope for an HLA allele only present in the source (Table 4), but in most cases, the peptide was likely to also be a target for shared HLA alleles with the recipient. Accordingly, overall there was limited evidence of reversion in the transmitted viruses in the recipient.
HCV-specific T cell response profile in local cohort of chronic HCV genotype 1a-infected subjects akin to pattern observed in the source
We compared the T cell responses we observed in the transmission pair to those we identified in our previous cross-sectional study of a local cohort of chronic HCV genotype 1a-infected subjects (23). Of the chronic infected subjects who carried HLA-A*02 (n = 63), seven subjects showed IFN-γ responses to at least one of the peptides in the panel tested in this study (Table 5). The IFN-γ ELISPOT results from these subjects showed that responses to a commonly targeted HLA-A*02-restricted T cell epitope (represented by the overlapping peptides at NS2821–829 and NS2822–831 in Table 2) often did not match the dominant contemporaneous autologous sequence, which carried the putative adapted amino acid (Table 5). These results are similar to what was observed in the source for the HLA-A*02-restricted NS5B2878–2886 epitope (DLPLIIQRL). In this example, the consensus sequence is present as the autologous sequence in both individuals, however, the T cell response was detected against the variant peptide with no T cell response observed to the peptide matching the autologous sequence (Table 5). We have previously predicted that in some instances the consensus sequence for the host population includes the adapted amino acid and this is consistent with the high frequency of HLA-A*02 in the population (12,25). These results further support the presence of viral adaptations in the transmitted source sequence.
Unless indicated, T cell epitope sequence from Pfafferott et al. (23) and restricted by HLA-A*02:01.
Population sequence obtained from local cohort of chronic genotype 1a subjects as described in Rauch et al. (25).
HLA-A2 subjects with chronic infection from Pfafferott et al. (23).
T cell epitope listed in IEDB. For peptides starting at NS2821/822 and NS5B2878, consensus includes putative adapted amino acid for HLA-A*02. Where available, bold indicates autologous viral sequence. N indicates no response and Y indicates response (≥25 SFU/million PBMCs).
PBMCs, peripheral blood mononuclear cells.
Discussion
In this study, high-resolution NGS and T cell immune response profiling methods were used to examine the early interplay between the virus and host immune response in a recent HCV transmission pair in which the source sequence was known and the sampling time points for the recipient were taken during acute HCV infection (with the first sampling time points likely to be within 6 weeks of infection). Despite the study of a single transmitter pair, the detailed analysis of the early events after transmission has revealed some important findings. The HCV strains that seeded infection in the recipient contained a number of likely adapted sites within the HCV genome for HLA-A*02; an allele shared by the source and recipient. The finding that T cell responses in the source were detected against peptides that did not represent the main circulating autologous virus supports the presence of adapted virus in the source. We found a similar pattern in a cohort of chronic HCV genotype 1a-infected subjects who were part of our cross-sectional study of anti-HCV immunity (23), in which viral adaptation is likely to exist. Importantly, responses against these peptides (adapted and nonadapted) were not detected in the recipient even though the T cell epitopes targeted by the source were restricted by shared HLA alleles. We favor the suggestion that the recipient may no longer be able to target these now adapted T cell epitopes. However, we cannot exclude that differences in the T cell response profile may also reflect the different TCR repertoire of the recipient relative to the source. Although it should be noted that some of the variant peptides tested differed from consensus at positions that are known to affect TCR recognition of the HLA-peptide complex.
The only response common to both the recipient and the source was against the peptide CVNGVCWTV of the NS31073–1081 T cell epitope, but for both subjects the dominant autologous virus was a different sequence (CINGVCWTV; common genotype 1A form). Although our own study did not find evidence of adaptation at the P2 position of this epitope (12), others have suggested variation at this site to be associated with HLA-A*02 (11). In this context, it is important to note that it is difficult to identify an HLA-associated viral adaptation using a population-based approach when the viral adaptation exists in the consensus sequence and is almost fixed at the population level, as the study requires a large number of subjects for sufficient statistical power. This T cell epitope has also been previously identified to elicit a T cell response in individuals due to past exposure to influenza A virus (35) and this may be an explanation for the positive IFN-γ T cell response detected in either subject, although further analysis is required for confirmation.
Interestingly, although an adapted transmitted founder virus is likely to have seeded infection in the recipient, the recipient went on to clear infection. We would have predicted that existing adaptation in the infecting virus might have negatively affected infection outcome. However, the identification of amino acid sites within potential T cell epitopes in the HCV genome that exhibited variation in the recipient relative to the source supports the presence of additional immune targets in the recipient. Due to limited PBMC availability, we were unable to test peptides that spanned these specific viral changes between the source and recipient and to assess their immunogenicity. Furthermore, the majority of peptides used in the IFN-γ ELISPOT screen covered variable areas in the HCV nonstructural proteins as the population-based genetic study on which many of the peptides were designed is based on the premise that viral escape to CD8+ T cell pressure is observed as HLA-associated viral polymorphisms (12,25). Accordingly, the peptide screen may have missed relevant HCV-specific T cell responses directed against conserved regions of the genome and/or the structural proteins of HCV that may have contributed to the resolution of the infection.
There are other host–viral factors that are likely to influence infection outcome and we did not test to determine if the recipient had neutralizing antibodies capable of clearance of infection. In addition, although the recipient carried the “risk” allele of IFNλ4, known to be a strong host predictor of infection outcome, we did not test for other genetic variants of genes involved in the innate immune response such as specific variants in the NK cell receptors (15).
In conclusion, this analysis suggests that adapted virus can be transmitted and maintained within a new host. The adaptive potential of the incoming viral sequence is likely to affect the host's targeted immune response against HCV during early infection and the effectiveness of this response will, in part, influence infection outcome as also recently shown for CD4+ T cell responses in another transmission case (7). Furthermore, transmission of preadapted virus in a new host is an important observation that, if widespread, will impact on the design of a protective HCV vaccine, as has been recently described for HIV (5). This will also need to be considered for viral adaptation sites to common HLA alleles present in the population that overlap with drug resistance mutations (13).
Footnotes
Acknowledgments
The authors thank the patients and clinical staff who participated in this study as well as their colleagues at the Institute for Immunology and Infectious Diseases, Murdoch University. They thank Kerensa McElroy for comments on previous versions of this article. This study was funded, in part, by the McCusker Research Foundation.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.