
Abstract
The Josephin domain-containing (JOSD) protein 1 (JOSD1) is recognized as one member of deubiquitinases (DUBs) due to its catalytic “Josephin” domain. However, the in vivo deubiquitinating activity of JOSD1 remains unidentified, and the biological functions of JOSD1 are largely unknown. In this study, we report that JOSD1 plays an important role in regulating type-I interferon (IFN-I)-mediated antiviral activity. JOSD1 physically interacts with SOCS1, which is an essential negative regulator of many cytokines signaling, and enhances SOCS1 stability by deubiquitinating K48-linked polyubiquitination of SOCS1. Furthermore, JOSD1 inhibits IFN-I-induced signaling pathway and antiviral response. Interestingly, during the early stage of viral infections, the levels of JOSD1 and SOCS1 undergo downregulation, which may facilitate activation of IFN-I signaling and efficient antiviral activity. Thus, our finding identified the first deubiquitinating substrate of JOSD1 and a novel biological function of JOSD1 and may provide a potential target for IFNs-based antiviral therapy.
Introduction
D
Among the five DUBs classes, MJD family is the smallest one which consists of four members: JOSD1, JOSD2, Ataxin3 (ATXN3), and Ataxin3-like (ATXN3L). Although they share a similar “Josephin” catalytic domain, only ATXN3 has been well studied so far. ATXN3 has been linked to the neurodegenerative disease, Machado–Joseph disease (MJD) (6). JOSD1, as a typical member of MJD family, contains only the highly conserved Josephin catalytic domain. However, the biological functions of JOSD1 remain largely unexplored. In particular, as a member of deubiquitinases, the in vivo deubiquitinating activity of JOSD1 remains unidentified.
Type-I interferon (IFN-I or IFNα/β), as a widely used antiviral drug in clinic, plays critical roles in regulating the immune defense against diverse pathogens (15). IFN-I family members bind to IFN-I receptors (IFNAR1 and IFNAR2) to induce tyrosine phosphorylation of Janus kinase family (JAK1 and TYK2). Activated Janus kinase family subsequently induces tyrosine phosphorylation and activation of the signal transducers and activators of transcription (STATs), including STAT1 and STAT2. Activated STAT1 translocates into the nucleus to induce the transcription of interferon-stimulated genes (ISGs). Finally, these ISGs perform a series of biological functions of IFN-I family, including antiviral defense, antiproliferation, and immunomodulatory effects (1,10).
Recently, lots of studies have demonstrated that excessive activated IFN-I signaling could play deleterious roles in autoimmune diseases (1,9), diverse models of endotoxic shock (19), and normal organismal homoeostasis (12). Therefore, how IFN-I signaling is negatively regulated by intracellular signaling has attracted more and more attention. For example, it has been reported that the suppressor of cytokine signaling protein 1 (SOCS1) is an important negative regulator of IFN-I signaling, which limits IFN-induced activation of JAK family (2), the adenosine deaminase acting on RNA 1 (ADAR1) plays essential roles in restricting IFN-I production and subsequent IFN-I signaling pathways (5,7).
In this study, we discover that JOSD1 physically interacts with SOCS1 and upregulates the level of SOCS1 protein. Furthermore, we found that JOSD1 increased SOCS1 protein stability through deubiquitinating K48-linked polyubiquitination of SOCS1. Critically, JOSD1 inhibited type-I IFN-induced STAT1 activation and downstream signaling pathway, and therefore restricted type-I IFN antiviral activity. Our findings identify the in vivo deubiquitinating activity of JOSD1 and reveal a novel mechanism, by which IFN signaling is negatively regulated by intracellular signaling.
Materials and Methods
Cell culture and transfection
Human embryo kidney 293T cells, human fibrosarcoma 2fTGH cells, and human colon cancer HCT116 cells were cultured at 37°C in humidified air containing 5% CO2. Cells were transfected with either LongTrans (Ucallm) or PEI (Polysciences) according to the manufacturer's instruction.
Antibodies, plasmids, and reagents
The following antibodies were used: Myc (Abmart), SOCS1 (Millipore), p-STAT1 (Cell Signaling), p-Tyk2 (Cell Signaling), JOSD1 (Santa Cruz), Ubiquitin (Santa Cruz), VSV-G (Santa Cruz), HA (Abcam), K48Ub (Cell Signaling), Flag (Sigma), β-Actin (Proteintech), and Alpha-Tubulin (Proteintech). FH-JOSD1 was from Dr. J. Wade Harper (Harvard Medical School, Addgene plasmids). Myc-SOCS1 was amplified by polymerase chain reaction (PCR) and subcloned into pcDNA3.1/Myc-His (Invitrogen). Flag-STAT1, Flag-Smurf1 (WT), HA-ub, ISRE-luciferase, and Renilla plasmids were as described previously (14). shJOSD1 constructs were purchased from Genechem (Shanghai, China). Myc-SOCS1 KR was generated by QuickChange site-Directed Mutagenesis Kit (Stratagene). Recombinant human IFNα was purchased from Peprotech. Cycloheximide (CHX) and Anti-Flag M2 Affinity Gel were purchased from Sigma.
Immunoblotting and immunoprecipitation
Immunoblotting and immunoprecipitation were performed as described previously (14). In brief, cells were lysed in NP-40 buffer containing 150 mM NaCl, 20 mM Tris-HCl (PH7.4), 1% Nonidet P-40, 0.5 mM EDTA, and 50 μg/mL PMSF. N-ethylmaleimide (10 mM) was added into the above lysis buffer when protein ubiquitination was detected. The cell lysates were centrifuged at 12,000 rpm for 15 min. The supernatants were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis. Proteins were identified using the Odyssey imaging system or detected by SuperSignal West Dura Extended Kits (Thermo Scientific). Immunoprecipitation was performed using specific antibodies overnight on a rotor at 4°C. Protein G agarose beads (No. 16-266; Millipore) were added into samples and incubated for an additional 3 h on a rotor at 4°C. The ImageJ program (
Reporter assays
Cells were transfected with the ISRE-luciferase and Renilla plasmids, together with specific constructs. After 36 h, cells were treated with IFNs overnight and then collected. The luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's protocol. Activity was assayed in three independent experiments and shown as the average mean ± standard deviation (SD).
Cycloheximide chase assay
The half-life of Myc-SOCS1 was determined by cycloheximide (CHX) chase assay as previously described (7).
RNA isolation and real-time PCR
Total RNAs were isolated using TRIzol reagent (Invitrogen). cDNA was synthesized by reverse transcription using oligo (dT) and subjected to quantitative real-time PCR with IFIT1, ISG54, and β-ctin primers in the presence of SYBR Green Supermix (BIO-RAD). The primer sequences are as described previously (14). The results were analyzed from three independent experiments and shown as the average mean ± SD. All statistical analyses were performed as described before (14).
Immunofluorescence microscopy
Cells infected with vesicular stomatitis virus with the green fluorescent protein (GFP) gene (VSV-GFP) at a multiplicity of infection (MOI) of 0.5 were subjected to analysis by immunofluorescence microscopy. In brief, cells with VSV-GFP infections were pictured with upright fluorescence microscope (Nikon 90i, Japan). Magnification was ×200.
Results
JOSD1 inhibits type I-IFN-mediated antiviral activity
Vesicular stomatitis virus (VSV) is widely used as a sensitive viral model to assess IFNs antiviral activity (4,13). We first analyzed the effect of JOSD1 overexpression on virus infections. To this end, human embryo kidney 293T cells, human fibrosarcoma 2fTGH cells, and human colon cancer HCT116 cells were transfected with or without Flag-HA-JOSD1 (FH-JOSD1) and then infected with VSV-GFP, a VSV construct with the GFP gene. One hour after infections, VSV RNAs levels were analyzed by quantitative real-time PCR. The results showed that overexpression of JOSD1 did not significantly affect the very early stage of VSV infections (Fig. 1A), indicating that JOSD1 does not influence viral entry.
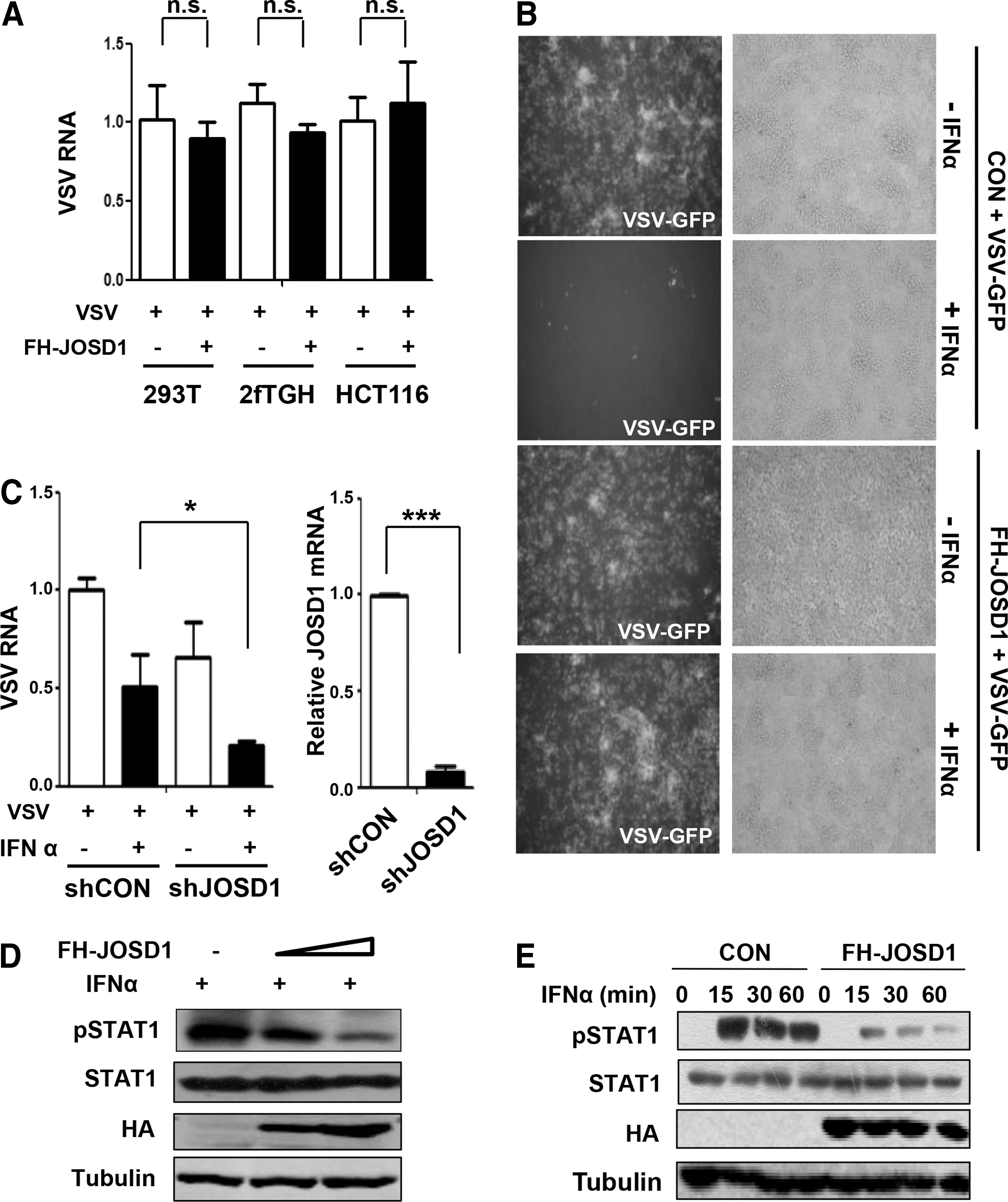
JOSD1 inhibits type I-IFN-mediated antiviral response.
Furthermore, the antiviral activity of type-I interferon (IFN-I) was analyzed by observing the GFP signal using fluorescence microscopy. As expected, IFN-I treatment remarkably inhibited VSV-GFP infections, as shown by a decreased GFP signal (Fig. 1B). Interestingly, overexpression of JOSD1 significantly inhibited IFN-I-mediated antiviral activity (Fig. 1B). To analyze the effect of endogenous JOSD1 on IFN-I antiviral activity, we used shRNA constructs against human JOSD1 (shJOSD1). 2fTGH cells transfected with control shRNAs (shCON) or shJOSD1 were treated with IFN-I, and then cells were infected with VSV. The result showed that knock-down of JOSD1 promoted IFN-I antiviral activity, as shown by decreased VSV viral RNAs in the shJOSD1 group (Fig. 1C), indicating that endogenous JOSD1 inhibits IFN-I antiviral activity. Based on the above data, we next tried to analyze whether JOSD1 is able to affect IFN-mediated STAT1 activation signaling. To this end, cells were transfected with FH-JOSD1 and then were treated with IFNα (1,000 U/mL) for 30 min. The result showed that IFNα-induced tyrosine 701 phosphorylation of STAT1 was remarkably downregulated (Fig. 1D). Similarly, the result was confirmed when cells were treated with IFNα for various time (Fig. 1E). Taken together, our data clearly demonstrate that JOSD1 is a negative regulator for IFN-I antiviral activity.
JOSD1 upregulates SOCS1 protein levels
To uncover the mechanisms by which JOSD1 regulates IFN-I antiviral activity, we first investigated whether JOSD1 affects the protein level of STAT1, which is recognized as a key signaling molecular in IFN-I signaling. We found that overexpression of JOSD1 did not noticeably affect STAT1 levels (Fig. 2A). Given that JOSD1 inhibits IFN-I antiviral activity, we speculate that JOSD1 may restrict IFN-I-mediated signaling through stabilizing certain negative regulators of IFN-I signaling. Smurf1 has recently been reported to be a negative regulator of IFNs signaling through ubiquitinating and degrading STAT1 protein (20). However, our data showed that JOSD1 overexpression did not obviously affect the protein levels of endogenous Smurf1 (Fig. 2B) and exogenous Flag-Smurf1 (Fig. 2C). Another negative regulator of IFN-I signaling, SOCS1, has been thought to be an important inhibitory protein for many cytokines signaling (2). Therefore, we examined the effect of JOSD1 on the protein level of SOCS1. Our data showed that overexpression of JOSD1 significantly upregulated endogenous SOCS1 (Fig. 2B) and exogenous SOCS1 (Fig. 2D) protein levels. These results suggest that JOSD1 positively regulates SOCS1 at the protein level.

JOSD1 interacts with SOCS1 and upregulates SOCS1 levels.
JOSD1 physically interacts with SOCS1
To confirm the influence of JOSD1 on SOCS1, we next examined whether JOSD1 is able to interact with SOCS1 in cells. 293T cells were cotransfected with FH-JOSD1 and Myc-tagged SOCS1 (Myc-SOCS1). By performing an immunoprecipitation assay, we found that FH-JOSD1 can interact with Myc-SOCS1 (Fig. 2E). Next, we tried to determine whether endogenous JOSD1 is capable of interacting with endogenous SOCS1. Our data clearly showed the interaction between endogenous JOSD1 and endogenous SOCS1 (Fig. 2F), which supports the possible effect of JOSD1 on SOCS1 protein levels. Collectively, we demonstrate that JOSD1 physically interacts with SOCS1 and positively regulates protein levels of cellular SOCS1.
JOSD1 deubiquitinates SOCS1
Given that JOSD1 can upregulate SOCS1 protein levels, we next questioned whether JOSD1, as a deubiquitinase, could deubiquitinate SOCS1 and affect SOCS1 protein stability. Until this point, the types of SOCS1 ubiquitination remain unknown. We found that SOCS1 can form polyubiquitination modification (Fig. 3A). Interestingly, we noticed that human SOCS1 gene harbors only one lysine (K). Furthermore, we demonstrated that the lysine 48 (K48)-linked polyubiquitination is the major form of SOCS1 ubiquitination (Fig. 3A).
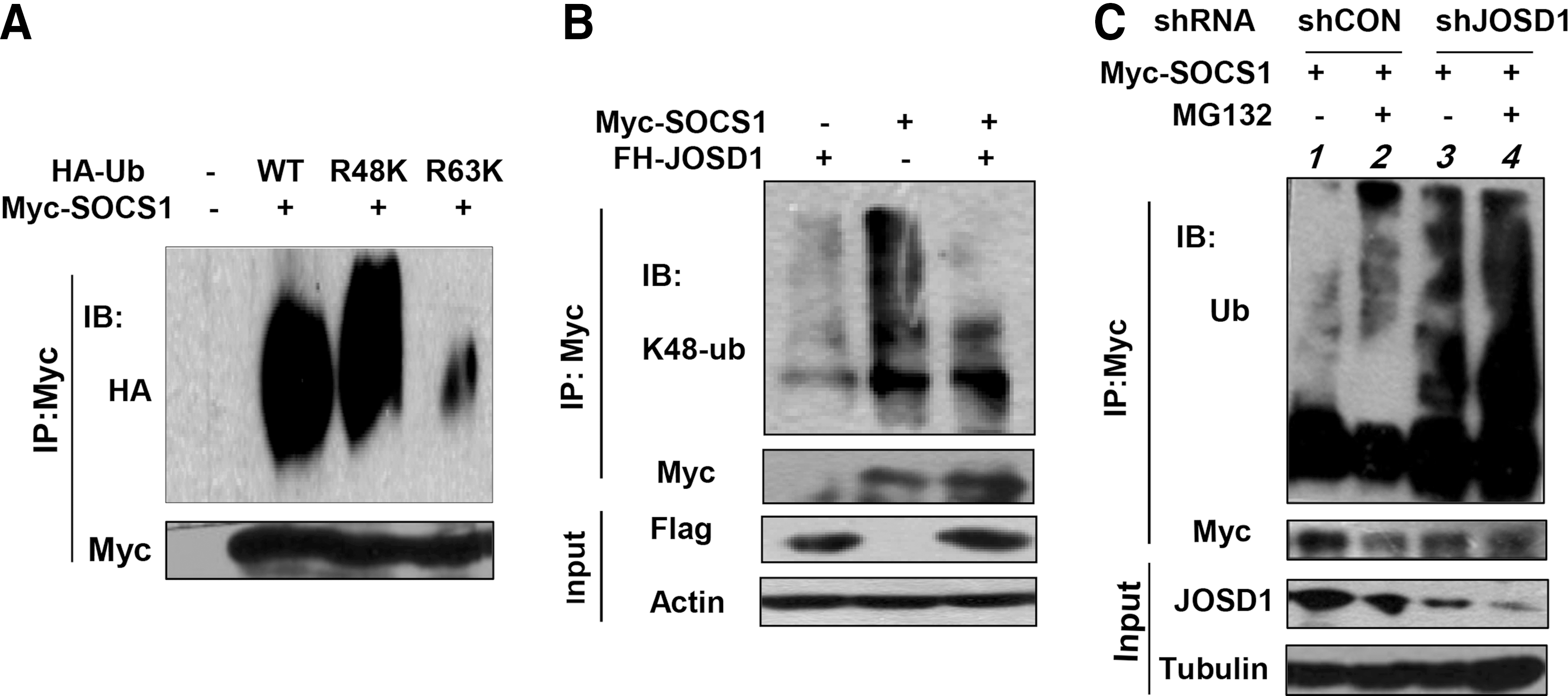
JOSD1 deubiquitinates SOCS1.
To examine the effect of JOSD1 on SOCS1 ubiquitination, cells were transfected with Myc-SOCS1, together with FH-JOSD1 or its empty vector. We found that overexpression of FH-JOSD1 significantly inhibited K48-linked polyubiquitination of Myc-SOCS1 (Fig. 3B). Conversely, when the proteasome inhibitor MG132 was used to block Myc-SOCS1 degradation, we found that ubiquitination of Myc-SOCS1 was obviously increased (Fig. 3C, lane 1 vs. lane 2), indicating that SOCS1 undergoes ubiquitination regulation in cells. When JOSD1 was knocked down in cells, polyubiquitination of SOCS1 was also remarkably increased (Fig. 3C, lane 1 vs. lane 3). These results demonstrate that JOSD1 can deubiquitinate K48-linked polyubiquitination of SOCS1.
JOSD1 stabilizes SOCS1 protein
Since JOSD1 can remove K48-linked polyubiquitination chains from SOCS1, we speculate that JOSD1 increases SOCS1 protein stability. The protein translation inhibitor cycloheximide (CHX) was used to inhibit protein synthesis. We find that the half-life of Myc-SOCS1 in cells is about 4 h (Fig. 4A). Furthermore, we observed that mutation of the only lysine on SOCS1 gene (KR) significantly enhanced SOCS1 protein stability (Fig. 4B), suggesting that SOCS1 proteins are tightly regulated by ubiquitination-dependent degradation in cells. Similarly, overexpression of JOSD1 also enhanced protein stability of endogenous SOCS1 (Fig. 4C). In summary, the deubiquitinase JOSD1 can remove K48-linked polyubiquitination of SOCS1, and therefore enhances SOCS1 protein stability.
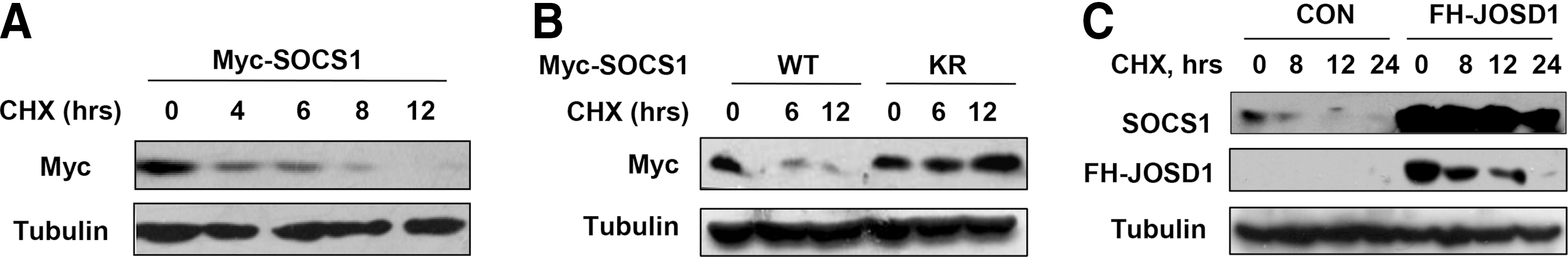
JOSD1 stabilizes SOCS1 protein.
Downregulation of both JOSD1 and SOCS1 levels during the early stage of viral infections
The aforementioned results demonstrate that JOSD1 is a positive regulator of SOCS1 protein levels. In conjunction with our previous results showing that JOSD1 inhibits IFN-I antiviral function, we speculate that JOSD1 could inhibit IFN-I-mediated signaling pathway. First, we want to know whether viral infections affect the levels of JOSD1 and SOCS1. Interestingly, we found that JOSD1 levels rapidly decreased during viral infections in both human fibrosarcoma 2fTGH cells (Fig. 5A) and HeLa cells (Fig. 5B).
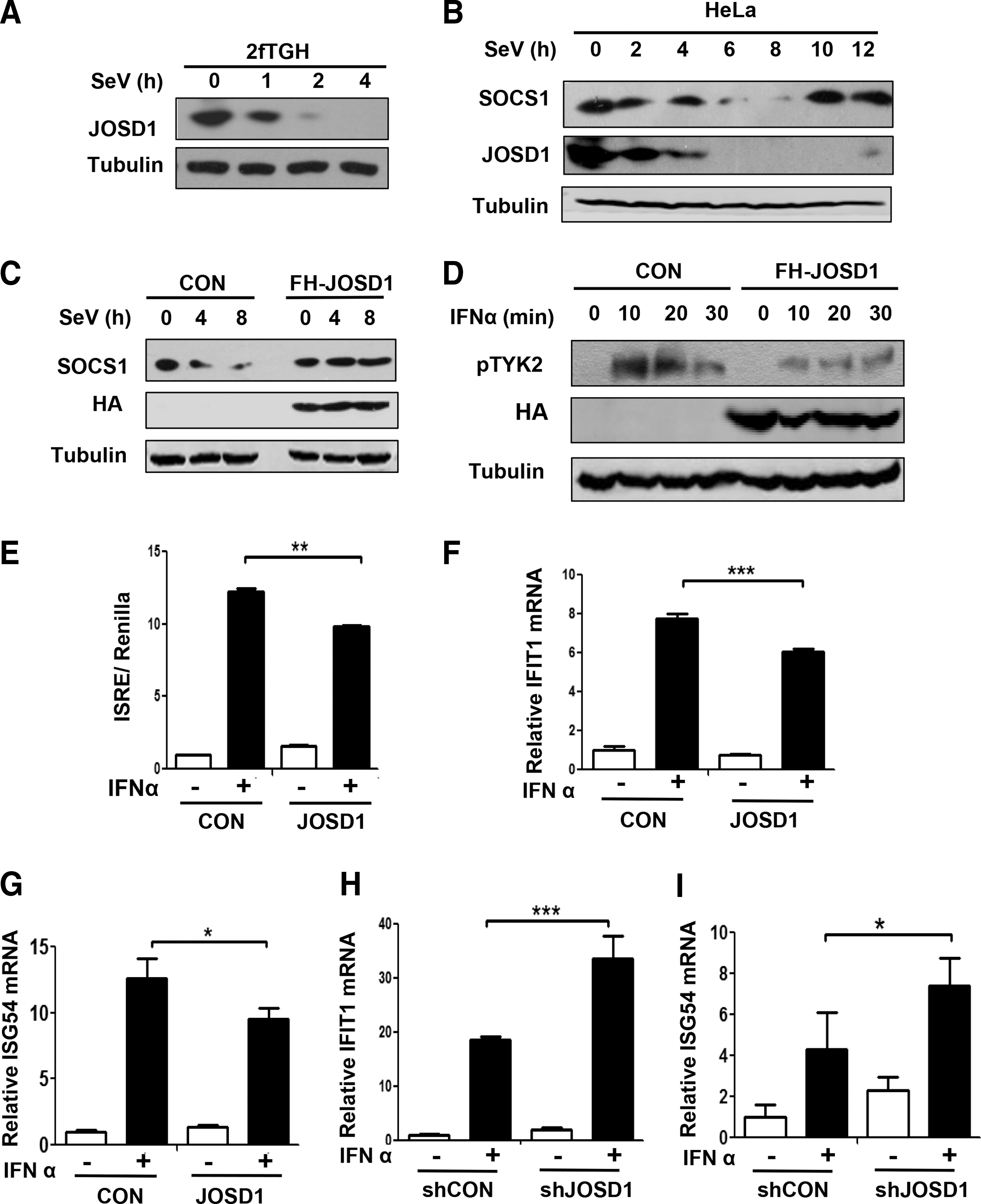
JOSD1 negatively regulates IFN-I-mediated signaling pathway via SOCS1.
Since we proved that JOSD1 is a deubiquitinase of endogenous SOCS1, we speculated that JOSD1 downregulation during viral infections could influence SOCS1 levels. Our data showed that SOCS1 levels were downregulated during the early stage of viral infections in HeLa cells (Fig. 5B). It is unexpected that SOCS1 levels are downregulated during viral infections. Given that viral infections induce IFN-I expression, and SOCS1 as an ISG gene can be upregulated by IFN-I, SOCS1 levels are predicted to be upregulated during viral infections. In this study, our findings may reveal an important mechanism by which SOCS1 levels undergo downregulation to facilitate activation of IFN-I signaling during the early stage of viral infections, since SOCS1 is a congenital inhibitor of IFN-I signaling. Next, we analyze whether SOCS1 downregulation could be blocked when cells were supplemented with enough JOSD1. To this end, cells transfected with FH-JOSD1 were infected with viruses. We found that JOSD1 overexpression blocked SOCS1 downregulation during the early stage of viral infections (Fig. 5C), indicating that JOSD1 can stabilize SOCS1 levels during viral infections.
JOSD1 negatively regulates IFN-I-mediated signaling pathway
Given that we demonstrate that JOSD1 positively regulates SOCS1 levels during viral infections, we speculate that JOSD1 is a negative regulator for IFN-I-mediated signaling pathway. It has been reported that the inhibitory effect of SOCS1 on IFNs signaling is through inhibition of tyrosine phosphorylation of TYK2 protein (11). Therefore, we further examined the level of tyrosine phosphorylation of TYK2 under IFN-I treatment. We found that overexpression of JOSD1 obviously inhibited tyrosine phosphorylation of TYK2 (Fig. 5D). This result is consistent with our above observation (Fig. 1D, E), which showed that overexpression of JOSD1 significantly inhibited Tyr701 phosphorylation of STAT1. Given that TYK2 phosphorylation induces STAT1 phosphorylation and activation, we think that JOSD1 can restrict STAT1 activation through promoting SOCS1-mediated inhibition on tyrosine phosphorylation of TYK2.
Based on the above results, we speculate that JOSD1 could negatively regulate IFN-I-mediated signaling pathway. The interferon-stimulated response element (ISRE) is widely used to analyze the promoter activities of ISGs. Therefore, ISRE is used to examine the magnitude of IFNs signaling (1). Cells were transfected with ISRE-luciferase constructs, together with FH-JOSD1 constructs, or empty vectors. The result showed that overexpression of JOSD1 obviously inhibits IFNα-induced ISRE-luciferase activities (Fig. 5E). To directly analyze the effect of JOSD1 on the activation of IFN-I-mediated genes, the mRNA levels of two representative ISGs, including IFIT1 and ISG54, were determined. Our data showed that overexpression of JOSD1 remarkably decreased mRNA levels of both ISGs under IFNα treatment (Fig. 5F, G). Conversely, knock-down of JOSD1 significantly increased mRNA levels of these two ISGs mediated by IFNα (Fig. 5H, I). Taken together, these results demonstrate that JOSD1 negatively regulates IFN-I-mediated signaling pathway.
Discussion
Deubiquitinases have been proved to be essential for regulating signaling pathways related to human diseases. Understanding the functions and deubiquitinating activities of deubiquitinases remains a major challenge. JOSD1 is a typical member of MJD deubiquitinase family. However, the biological functions of JOSD1 remain largely unexplored. In this study, we reveal that JOSD1 is a negative regulator for IFN-I mediated antiviral response (Fig. 1). Our study establishes a link between the deubiquitinase JOSD1 and IFN-mediated innate antiviral immunity. Therefore, our finding indicates that JOSD1 could be a potential target for improving IFNs-based antiviral therapy.
So far, only a few studies reported the in vitro deubiquitinating effect of JOSD1 (17). The in vivo deubiquitinating activity of JOSD1 remains unknown. Our study reveals that JOSD1 can deubiquitinate SOCS1 (Fig. 3) and enhances protein stability of cellular SOCS1 (Fig. 4). SOCS1 is an important negative regulator of many cytokines (including IFNs) signaling. It is less clear how SOCS1 protein is regulated by deubiquitination modification. Therefore, our study is the first report that JOSD1 possesses the in vivo deubiquitinase activity and uncovers the deubiquitination regulation of SOCS1 protein.
An interesting study showed that JOSD1 can regulate membrane dynamics and cell motility. However, this biological function is independent of the deubiquitinase activity of JOSD1 (17). In this study, we identify SOCS1 as the in vivo deubiquitinating substrate of JOSD1. Our data demonstrate that JOSD1 positively regulates SOCS1 levels by removing K48-linked polyubiquitination of SOCS1 (Fig. 3) and therefore inhibits IFN-I-mediated signaling pathway and antiviral function (Fig. 1). Thus, our finding reveals a novel biological function of the deubiquitinase JOSD1.
In summary, this study demonstrates that JOSD1 physically interacts with and deubiquitinates SOCS1, thereby enhancing SOCS1 protein stability and upregulating SOCS1 protein levels. Importantly, JOSD1 inhibits IFN-I-induced signaling pathway and restricts IFN-I-mediated antiviral response during viral infections. This finding uncovers the in vivo deubiquitinating activity of JOSD1 and reveals that the deubiquitinase JOSD1 is a negative regulator of IFN-I antiviral response.
Footnotes
Acknowledgments
We thank Dr. Chen Wang from Shanghai Institutes for Biological Sciences of Chinese Academy of Sciences in China, and Dr. Chunsheng Dong from Soochow University for important plasmids or viruses. This work was supported by the National Natural Science Foundation of China to H.Z. (31370873, 31570865) and to T.G. (31501139); the Program of 1000 Young Talents to H.Z., Jiangsu Provincial Distinguished Young Scholars to H.Z. (BK20130004), Changjiang scholars and Innovative Research Team in University of Ministry of Education of China (PCSIRT-IRT1075), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Author Disclosure Statement
No competing financial interests exist.