
Abstract
The effect of the current influenza vaccine, an inactivated virus vaccine administered by subcutaneous/intramuscular injection, is limited to reducing the morbidity and mortality associated with seasonal influenza outbreaks. Intranasal vaccination, by contrast, mimics natural infection and induces not only systemic IgG antibodies but also local secretory IgA (S-IgA) antibodies found on the surface of the mucosal epithelium in the upper respiratory tract. S-IgA antibodies are highly effective at preventing virus infection. Although the live attenuated influenza vaccine (LAIV) administered intranasally can induce local antibodies, this vaccine is restricted to healthy populations aged 2–49 years because of safety concerns associated with using live viruses in a vaccine. Instead of LAIV, an intranasal vaccine made with inactivated virus could be applied to high-risk populations, including infants and elderly adults. Normally, a mucosal adjuvant would be required to enhance the effect of intranasal vaccination with an inactivated influenza vaccine. However, we found that intranasal administration of a concentrated, whole inactivated influenza virus vaccine without any mucosal adjuvant was enough to induce local neutralizing S-IgA antibodies in the nasal epithelium of healthy individuals with some immunological memory for seasonal influenza viruses. This intranasal vaccine is a novel candidate that could improve on the current injectable vaccine or the LAIV for the prevention of seasonal influenza epidemics.
Introduction
Properties of influenza
I
At present, influenza A(H1N1)pdm09 viruses, influenza A(H3N2) viruses, and influenza B viruses cocirculate worldwide (30,134,135). The A(H1N1)pdm09 virus caused the 2009 pandemic and, subsequently, replaced the A(H1N1) virus that had predominated before 2009 [hereafter, A(H1N1)pdm09 is referred to as A(H1N1) in this review] (27,44,85,90,115).
The clinical signs and symptoms associated with seasonal influenza are high fever (up to 40°C), chills, cough, sore throat, runny or stuffy nose, muscle or joint aches, headache, and fatigue. In health adults, influenza symptoms begin ∼2 days (1–4 days) after exposure to virus and abate within a week without any medical treatment. Serious influenza complications (such as virus/bacteria coinfection, secondary bacterial pneumonia, myocardial dysfunction, myositis, multiorgan failure, and influenza-associated encephalopathy) are observed in young children, elderly adults (more than 65 years old), pregnant women, and immunosuppressed individuals (30,118,126,134,135). The World Health Organization (WHO) estimates that, globally, three to five million people experience severe illness and that 250,000 to 500,000 die in annual influenza epidemics (135). Currently, medical treatments for the prevention and treatment of influenza include antiviral agents and prophylactic vaccination.
Purpose of this review
The current percutaneous influenza vaccine is effective for the reduction of morbidity and mortality after influenza virus infection (135), but is less effective for the prevention of viral infection. This contrasts with the immunity acquired after natural infection, which prevents new viral infections. This protection at the site of infection is due to secretory IgA (S-IgA) antibody responses on the surface of nasal mucosa, which protect against both homologous and heterologous viruses (23). Although intranasal administration of live attenuated influenza vaccine (LAIV), which mimics natural infection, can also induce local S-IgA antibodies (9,17,54), its efficacy against the A(H1N1) virus appeared lower compared with the percutaneous influenza vaccine. An alternative strategy to induce S-IgA antibodies would be the intranasal administration of an inactivated influenza vaccine (IIV) combined with a mucosal adjuvant as an immunostimulator to enhance the immunogenicity of the IIV. Materials imitating pathogen-associated molecular patterns (PAMPs) sensed by pattern-recognition receptors could be effective adjuvants (34,73). For one, synthetic double-stranded RNA (dsRNA), which likely imitates intermediates produced during viral replication, induces cross-protective local S-IgA antibodies that are effective against both homologous and heterologous challenges.
To improve the current influenza vaccine and develop a more effective one, we should seek to understand the innate and adaptive immune responses to influenza virus infection, the effects of the currently available percutaneous IIV and LAIV, and the issues with these vaccines that need to be addressed. With these considerations in mind, this review article focuses on the usefulness of intranasal vaccination with IIVs in the presence of mucosal adjuvant as an alternative to LAIV. We also summarize results from our recent study of nonadjuvanted intranasal vaccination with a whole inactivated virus vaccine (WIVV).
Immune Responses Against Influenza Virus Infection
During natural influenza virus infections, the mucosa of the upper respiratory tract is not only the initial site of infection but also the first line of defense. Influenza virus that passes the first, primitive defense line (the mucin layer, ciliary action, and protease inhibitors) is then recognized by several classes of pattern recognition receptors, including Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), and NOD-like receptors (NLRs), expressed on innate immune cells such as dendritic cells (DCs) and macrophages.
Influenza A and B viruses have a negative-sense, single-stranded, and eight-segmented RNA genome (135). During replication, these viruses generate dsRNA intermediates. Single-stranded virus RNAs (vRNAs) are recognized by TLR7 coupled with the cytosolic adaptor molecule MyD88 in endolysosomal compartments of plasmacytoid DCs (34,36,76). The TLR7 signaling pathway induces production of vast amounts of interferon-α (IFN-α) (56,69). Separately, dsRNA intermediates are recognized by TLR3 coupled with the cytosolic TRIF protein in endolysosomal fraction of alveolar and bronchial epithelial cells or conventional DCs (4,34,50,55) and enhance the production of IFN-β, as well as pro-inflammatory cytokines (37,137). Moreover, the cytosolic sensor RIG-I, a RLR, recognizes vRNAs bearing a 5′-triphosphate in infected cells (98,102), inducing the production of type I IFN and pro-inflammatory cytokines (70). IFN-α/β production enhances the expression of IFN-stimulated genes with antiviral activity (PKR, RNaseL, Mx proteins, and so on) (100,135). A member of the NLR family, NLRP3, is also activated after influenza virus infection, converting pro-IL-1β and pro-IL-18 into mature pro-inflammatory cytokines (IL-1β and IL-18) (5,59,60,125).
In addition to the cytokine profiles induced by influenza virus infection, naive CD4+ T lymphocytes are activated by viral antigens presented on antigen-presenting cells (APCs, mainly cDCs). These then differentiate into corresponding effector helper T cells that participate in the induction and control of adaptive immune responses (82). Intact viruses or viral antigens released from infected cells are incorporated by APCs through endocytosis. Viral peptides are then usually presented by major histocompatibility complex class II (MHC II) molecules to the CD4+ T lymphocytes in peripheral secondary lymphoid tissues (130). The recognition of antigenic peptides on MHC II molecules by the T cell receptor in concert with secondary stimulation signaling activates effector CD4+ T lymphocytes. Naive B cells take up viral antigens binding to B cell receptors and present degradation products on MHC II molecules. With the help of activated follicular helper T (Tfh) cells, these B cells then differentiate into plasma cells producing class-switched and affinity-matured antibodies (22,38,78,111). Concurrently, a portion of the degradation products is cross-presented by MHC I to CD8+ T lymphocytes (21,130), resulting in the activation and differentiation of naive CD8+ T cells into cytotoxic T lymphocytes (CTLs) (10), which eliminate virus-infected epithelial cells (57,66,89,127).
The most characteristic feature of the adaptive immune response against influenza virus infection is the induction of S-IgA antibodies on the mucosa of the upper respiratory tract in addition to the induction of systemic IgG antibodies (23). The viral surface glycoproteins, HA and NA, are major antigens targeted by antibodies (135). HA-specific antibodies are primarily responsible for the prevention of infection by neutralizing influenza viruses, while NA-specific antibodies likely modify disease severity by restricting virus replication (65,135). IgA class switching is modulated by the induction of TGF-β (20,26), and high numbers of plasmablasts producing IgA antibody are generated under TGF-β and IL-21 signaling promulgated by Tfh cells (38).
IgA antibodies are joined by a J-chain and bind to a polymeric Ig receptor (pIgR) located on the basolateral surface of epithelial cells. The antibodies are then carried to the apical surface by transcytosis where they combine with the extracellular region of pIgR, which is cleaved as a secretory component by a specific protease to generate S-IgA antibodies (23,117). S-IgA antibodies are actively transferred to the mucus by transcytosis, while IgG antibodies migrate from the serum to the mucus by diffusion (11,12,23,80,81). In the upper respiratory tract, S-IgA antibodies prevent new viral infection with the support of IgG antibodies, which neutralize newly synthesized viruses (64,103). In contrast, in the lung, IgG antibodies predominate (64,95,101,103). The unique feature of S-IgA antibodies is their cross-protective effect against antigenically heterologous viruses. Indeed, blocking the transcytosis of IgA antibodies in pIgR-knockout mice resulted in a failure to protect against challenge with a heterologous influenza virus (11,12).
Currently Available Influenza Vaccines
IIV and LAIV
Worldwide, subcutaneous or intramuscular injectable IIVs are currently used to prevent epidemics caused by seasonal influenza viruses (30,132,134,135). A WIVV, chemically inactivated with formalin or β-propiolactone, was the first influenza vaccine. Since the 1970s, however, an ether-disrupted split IIV, mainly consisting of HA antigen, has been used because it shows nearly equivalent immunogenicity to the WIVV in primed populations (132). This IIV induces systemic IgG antibodies and effectively reduces the morbidity and mortality caused by influenza. The IIV can be applied to infants older than 6 months, adults, and the elderly (a high dose formulation is licensed for use in persons older than 65 years) (47).
At present, intranasal vaccination with a LAIV is also available as FluMist® or Fluenz® in the United States and the European Union, respectively. The advantage of an intranasal LAIV is that it mimics the natural route of infection and so induces production of S-IgA antibodies in the upper respiratory tract, preventing virus from infecting the nasal epithelium, as well as CTL responses and serum IgG antibody responses (9,17,54). The LAIV is only authorized for use in healthy children and adults 2–49 years of age because of safety concerns (47,108). This means that the groups at greatest risk for influenza complications, namely infants and the elderly, are unfortunately unable to receive the LAIV.
Depending on global surveillance of influenza virus circulation in humans, the WHO Global Influenza Surveillance and Response System updates its recommendation for the virus strain composition of the seasonal influenza vaccine twice a year (134). Before the 2013/14 influenza season, the WHO recommended three influenza virus strains [one strain of A(H1N1), one strain of A(H3N2), and one influenza B strain from two different lineages]. Currently, however, the recommendations include one more influenza B virus strain derived from the other lineage (30,47,134), because predictions of the circulating influenza B virus lineage were correct in only 5 of 10 seasons from 2001/02 to 2010/11 (7).
Vaccine effectiveness of LAIV
The LAIV licensed as a trivalent formulation (LAIV3) in the United States in 2003 is currently available as a quadrivalent formulation (LAIV4) among healthy individuals aged 2–49 years. Several clinical studies comparing the effectiveness of LAIV3 to that of IIV3 revealed that, although the efficacy of IIV3 was similar or slightly higher in healthy adults aged 17–49 years (8,79,91,92,128,131), the efficacy of LAIV3 was superior in children 6 months to 18 years old (8,14,18,19,40,99). In June 2014, the US Advisory Committee on Immunization Practices (ACIP) recommended the use of LAIV for healthy children aged 2–8 years old without contraindications or precautions. However, the effectiveness of LAIV4 against A(H1N1) strains in children aged 2–17 years old decreased during the 2013/14 season (25,31,42) (Table 1).
CI, confidence interval; IIV, inactivated influenza vaccine; LAIV, live attenuated influenza vaccine; VE, vaccine effectiveness.
One possible reason thought to explain this was the instability and low infectivity of a live attenuated A/California/7/09 (H1N1) virus isolated during the early stage of the 2009 pandemic (25). The A/California/7/09 virus possesses a glutamine residue (E) at the 374th position of HA (position 47 in HA2 numbering), while more recent isolates of the A(H1N1) virus circulating since July 2009 contain a lysine residue (K) at this position. This E47K substitution improves viral membrane fusion activity, acid and thermal stability, and infectivity in vivo (32). Based on this result, the A/California/7/09 strain was exchanged for a more recent isolate, A/Bolivia/559/13, in the LAIV4 for the 2015/16 season. During the 2015/16 season, although the effectiveness of the LAIV4 was statistically moderate against A(H1N1) in children, it was still lower compared with the subcutaneous inactivated influenza virus vaccine (47,88,96,97) (Table 1). In view of these observations, the ACIP withdrew the recommendation to use LAIV in the United States for the 2016/17 season in June 2016 (29).
Recently, Ambrose et al. posed the following four hypotheses to explain the low vaccine effectiveness of the LAIV against A(H1N1): (i) poor thermal stability of the vaccine; (ii) low replicative fitness of A(H1N1); (iii) viral interference in the quadrivalent formulation; and (iv) suppressed replication under preexisting influenza virus immunity (6). The first possibility was discarded in a report on vaccine effectiveness during the 2015/16 season (47). Regarding poor replicative fitness, Ambrose et al. revealed that both the A/California/7/09 and A/Bolivia/559/13 strains showed not only reduced replication in a human alveolar cell line and in primary nasal epithelium air-liquid cultures but also weak binding to α2,6-linked sialic acid receptors for human influenza viruses (6). If replication of A(H1N1) in the epithelium of the upper respiratory tract in humans is insufficient, viral interference between the vaccine strains contained in the LAIV could explain the vaccine's poor effectiveness against A(H1N1).
Low effectiveness against A(H1N1) was observed during the 2010/11 season (31) (Table 1) when A(H3N2) viruses predominated in the United States, with only local circulation of A(H1N1) viruses in some regions of the United States (28). Since the first use of LAIV4 coincided with the first dominant circulation of A(H1N1) since the 2009 pandemic, the change from a trivalent to a quadrivalent LAIV formulation was suspected to be a main reason for its poor performance against A(H1N1). In a cohort study conducted in Finland, the effectiveness of LAIV4 against influenza A virus in 2-year-olds who had previously received the seasonal IIV3 was higher than it was among those not previously vaccinated (88). This result suggested that at least preexisting influenza immunity acquired by IIV might have no effect on vaccination with LAIV4. At present, the exact reason for low effectiveness against A(H1N1) has not been clearly elucidated.
The four hypotheses described above largely derive from properties of the LAIV strains; that is, the efficacy of intranasal LAIV administration relies on the processes of infection and replication in nasopharyngeal epithelial cells, which may depend a great deal on the nature of the individual LAIV strains.
Antigenic changes between wild-type virus and vaccine strains
For the production of influenza vaccine, high-yield reassortant vaccine strains are generated and propagated in embryonated hen eggs. A reassortant vaccine strain bearing HA and NA derived from a prevalent wild type is prepared by simultaneously injecting eggs with a wild-type virus recommended by the WHO and an egg-adapted donor virus, such as the A/Puerto Rico/8/34 or the B/Lee/40 virus. Recently, it was suspected that the antigenicity of the vaccine might be different from that of the original wild-type virus in the case of the A(H3N2) and influenza B viruses (71,104,110,112,113). Amino acid substitutions in the HA of influenza B virus that result from egg adaptation are accompanied by the loss of the N-linked glycosylation site, causing a remarkable alteration of antigenicity (104,110,112). Furthermore, egg adaptation of contemporary A(H3N2) viruses introduces amino acid mutations in the receptor-binding site of their HA molecule, causing antigenic change relative to wild-type viruses (71,113). These antigenic alterations in vaccine strains of A(H3N2) and influenza B viruses might contribute to the reduction of vaccine effectiveness.
Usefulness of Intranasal IIV
Despite its limitations (6), LAIV is superior to conventional influenza vaccines in terms of the induction of mucosal immunity (9,17,54). Another vaccination method that induces mucosal immunity is intranasal vaccination with an inactivated influenza virus vaccine.
There have been numerous attempts to develop an inactivated vaccine that can be administered through the mucosal route. Since an inactivated vaccine alone is insufficient to elicit a full immune response at the mucosal surface because of its weak immunogenicity, an adjuvant is required to enhance the immune response. Traditionally, bacterial toxins (cholera toxin [CT] and heat-labile enterotoxin [LT]), those B subunits (CTB and LTB), or CTB containing a trace amount of whole toxin (CTB*) have been used to enhance mucosal immune responses after intranasal vaccination. In the upper respiratory tract, these mucosal adjuvants induce effective cross-protection against different viruses of the same subtype (121 –124). This strong cross-protection was provided mainly by S-IgA antibodies, whereas weak cross-protection in the lower respiratory tract was provided by IgG antibodies (39,121,124). An inactivated virosomal-subunit influenza vaccine adjuvanted with LT was licensed in Switzerland (Nasalflu) and made available for the 2000/01 influenza season. This Nasalflu was the first licensed intranasal influenza vaccine in the world. However, the incidence rate of Bell's palsy was higher among recipients of Nasalflu than among IIV recipients (84). This incident was presumably associated with the use of LT as a mucosal adjuvant (75,84). In response, this vaccine was removed from clinical use.
Many subsequent attempts have been made to identify an alternative adjuvant for intranasally administered inactivated vaccines. There are many candidates, including synthetic dsRNAs (58,61,62), CpG (13), chitin or surf clam microparticles (52,63), α-galactosylceramide (67), poly(gamma-glutamic acid) nanoparticles (94), and monophosphoryl lipid A (16). Of these potential adjuvants, synthetic dsRNAs are particularly interesting. Synthetic dsRNAs likely mimic dsRNA intermediates produced during influenza virus replication that are sensed by TLR3 or cytosolic receptor MDA5 (4,68).
Intranasal administration of an IIV prepared from A/Puerto Rico/8/34 (H1N1, designated A/PR8) combined with synthetic dsRNA poly(I:C) conferred protection against homologous A/PR8 not only in the upper respiratory tract but also in the lung (62). It also successfully induced strong anti-HA S-IgA antibodies in nasal washes and IgG antibodies in serum samples. Against challenge with A/PR8 virus into the nasal cavity, intranasal administration of each IIV derived from variant viruses of A(H1N1) showed complete protection with increases of A/PR8 HA-reactive nasal S-IgA antibodies, while nasal vaccination with IIV derived from an A(H3N2) virus, or influenza B viruses, provided little or no protection with limited production of S-IgA antibodies against A/PR8 HA (62).
While effective as an adjuvant, there are problems with poly(I:C). The poly(I:C) molecules can elongate by self-annealing, causing toxicity (77). Poly(I:C) also induces side effects in humans, including renal failure and hypersensitivity (106). Another synthetic dsRNA is PolyI:PolyC12U, which contains uridine residues and generates unique “mismatched” dsRNAs capable of undergoing accelerated hydrolysis (116). Mice vaccinated either intranasally or subcutaneously with a WIVV of a highly pathogenic avian influenza A(H5N1) virus, A/Vietnam/1194/2003 (designated as A/VN), in the presence of PolyI:PolyC12U survived challenge with the homologous A/VN virus. The propagation of virus in the nasal cavity was completely abolished in intranasally vaccinated mice, but not in those subcutaneously vaccinated (58). Against lethal challenge with variant A(H5N1) viruses (A/Hong Kong/843/97 or A/Indonesia/6/05 virus, designated as A/HK or A/IN, respectively), mice that received an A/VN WIVV with PolyI:PolyC12U intranasally exhibited higher survival rates than those subcutaneously vaccinated and exhibited significant reductions in viral loads in the nasal cavity (58). Furthermore, intranasal, not subcutaneous, administration of the IIV3 for the 2005/06 season with PolyI:PolyC12U improved survival and partially reduced nasal virus shedding during infection of heterologous highly pathogenic avian influenza A(H5N1) viruses (A/VN, A/HK, or A/IN) (61).
These results clearly indicate that intranasal administration of an IIV together with synthetic dsRNA, poly(I:C) or PolyI:PolyC12U, can enhance cross-protection by inducing S-IgA antibodies on the surface of mucosal epithelium in the upper respiratory tract.
For the global application of an effective vaccine, it is desirable to induce strong immune responses using a minimal vaccine dose. The enhancement of immune responses by the addition of an adjuvant might lead to dose sparing. Numerous studies on innate immune systems indicate that PAMPs and endogenous damage-associated molecular patterns recognized by pattern-recognition receptors could be adjuvant candidates (34,73). Moreover, simultaneous or sequential stimulation through MyD88-dependent and TRIF-dependent signaling pathways by their respective ligands induces a synergistic increase in cytokine production from mouse bone marrow-derived DCs or macrophages (15). Consistent with this, intranasal vaccination of an IIV of A/PR8 in the presence of poly(I:C) and zymosan, a cell wall extract from Saccharomyces cerevisiae, synergistically enhanced the production of protective local S-IgA and systemic IgG antibodies in mice (1). Although zymosan recognition by Dectin-1 and TLR2 results in the synergistic activation of DCs or macrophages through both the Dectin-1/syk and TLR2/MyD88 signaling pathways (24,35,43,49,53,105,107,114), the combination of zymosan with poly(I:C) was presumed to enhance an additional signaling pathway through TLR3/TRIF (1).
Thus, combinations of ligands on different innate immune sensors might be useful as adjuvants because they synergistically induce cross-protective antibodies after intranasal vaccination. This could be a promising strategy not only for dose sparing of antigen but also to reduce the total amount of adjuvant used.
Intranasal Vaccination with WIVV
Although intranasal vaccination with IIV in the presence of an appropriate mucosal adjuvant significantly induces cross-protective S-IgA antibodies in the nasal cavity, there is currently no mucosal adjuvant authorized for practical use. We know, however, that a WIVV is more immunogenic than a split-virus vaccine such as IIV (33) and that intranasal administration of a WIVV, not a split-virus vaccine, could induce a broad spectrum of heterosubtypic immunity against influenza virus infection (120). Koyama et al. revealed that the higher immunogenicity of WIVV depends on intrinsic vRNAs recognized by TLR7 and that this format of the vaccine is superior for its priming effect in naive subjects (72). Together, these reports suggest that a WIVV could induce more effective immune responses than a split vaccine after intranasal vaccination in healthy adults.
Studies conducted on humans indicated that intranasal vaccination with WIVVs increased the production of both local HA-specific IgA antibodies and serum hemagglutination inhibiting (HI) antibodies (45,46,83). In these reports, levels of local HA-specific antibodies were estimated by the enzyme-linked immunosorbent assay (ELISA); however, the neutralizing activity of the local antibody is more physiologically important in regards to protection against influenza virus infection.
Considering this, we estimated neutralization or HI antibody responses in serum and nasal wash samples after intranasal administration of a split vaccine or a WIVV to healthy adult volunteers (2,3). Each vaccine dose contained 45 μg of HA and was prepared from a A(H3N2) vaccine strain, which corresponded to threefold higher amounts of the representative strain than in seasonal IIVs. Intranasal vaccination with a monovalent split vaccine of A/Uruguay/716/07 virus was performed five times at 3-week intervals in five individuals (2). Intranasal vaccination with a monovalent WIVV of A/Victoria/210/09 virus was performed twice, 3 weeks apart, in 50 healthy adult participants (3). Remarkably, neither intranasal vaccination included a mucosal adjuvant. When a split vaccine was chosen as the antigen in an intranasal vaccination, four doses were required to induce a 2.5-fold increase in geometric mean titers (GMTs) of serum neutralizing antibodies (2). By contrast, only two doses of an intranasal WIVV were needed to induce more than a 2.5-fold increase of GMT (3). These results suggest that WIVV is superior to a split product. Local antibody responses were measured in concentrated nasal wash samples (1 mg/mL total protein) that contained ∼1/10 of the IgA and IgG antibodies found in undiluted nasal mucus (2,74).
In our human study of intranasal vaccination with a WIVV of A(H3N2) (3), the ratios of GMTs for serum HI and neutralization titers against homologous A/Victoria/210/09 virus were 4.3 and 8.0, respectively, among 46 out of 50 volunteers under 60 years of age (Table 2). Consistent with serum antibody responses, HI and neutralization antibody responses in standardized nasal wash samples showed 3.1- and 5.9-fold increases, respectively (Table 2). Serum neutralization titers correlated well with HA-specific IgG antibody titers estimated by ELISA (r = 0.778, p < 0.0001), but not with IgA antibody titers. Neutralization titers in nasal wash were correlated with HA-specific IgA antibody titers, but not with IgG antibody titers (r = 0.473, p < 0.001). Cross-reactive HI and neutralization titers to the variant A/Sydney/05/97 (H3N2) virus were induced ∼1.5-fold after two doses of intranasal vaccination (Table 2). Nasal HI and neutralization titers against the variant virus increased 1.6- and 2.1-fold, respectively (Table 2).
Reproduced from Ainai et al. (3). Intranasal vaccination with a monovalent WIVV of A/Victoria/210/09 (H3N2) was performed twice on week 0 and 3. Serum and nasal wash were collected on week 0, 3, and 6 for the evaluation of antibody responses.
Serum samples were treated with the receptor destroying enzyme [RDE(II); Denka Seiken, Tokyo, Japan] and finally diluted 1:10. Serial twofold dilutions from 1:10 were used for HI and neutralization assay. Serum antibody titers of <1:10 were considered negative and arbitrarily assigned a titer of 1:5.
Nasal antibody titers were determined using concentrated nasal wash samples containing 1 mg/mL total protein, corresponding to 10-fold diluted IgA and IgG antibodies found in undiluted nasal mucus. Standardized nasal wash samples were treated with RDE(II) and finally diluted 1:2, corresponding to 1:20 diluted nasal mucus. Serial twofold dilutions from 1:20 as nasal mucus were used for HI and neutralization assay. Nasal antibody titers of <1:20 were considered negative and arbitrarily assigned a titer of 1:10.
GMT, geometric mean titer; HI, hemagglutination inhibiting; WIVV, whole inactivated virus vaccine.
In addition, we clearly identified that large polymeric S-IgA antibodies play crucial roles in the protective immunity against influenza virus infection in the nasal cavity (119). Although the existence of highly polymerized S-IgA antibodies had been suggested previously (133), their formulation and mechanism had not been elucidated. In our study, nasal wash fluids were collected from participants who had received intranasal vaccination with a WIVV, and S-IgA antibodies were subsequently purified and separated by gel filtration chromatography. High-speed atomic force microscopy revealed that human nasal S-IgA molecules consist of at least five quaternary structures, including monomeric, dimeric, trimeric, and tetrameric structures, as well as a polymeric form larger than the tetramer structure (Fig. 1). The neutralizing activity was highest in the fraction of highly polymerized S-IgA antibody and decreased in the order of trimeric/tetrameric, dimeric, and monomeric fraction.
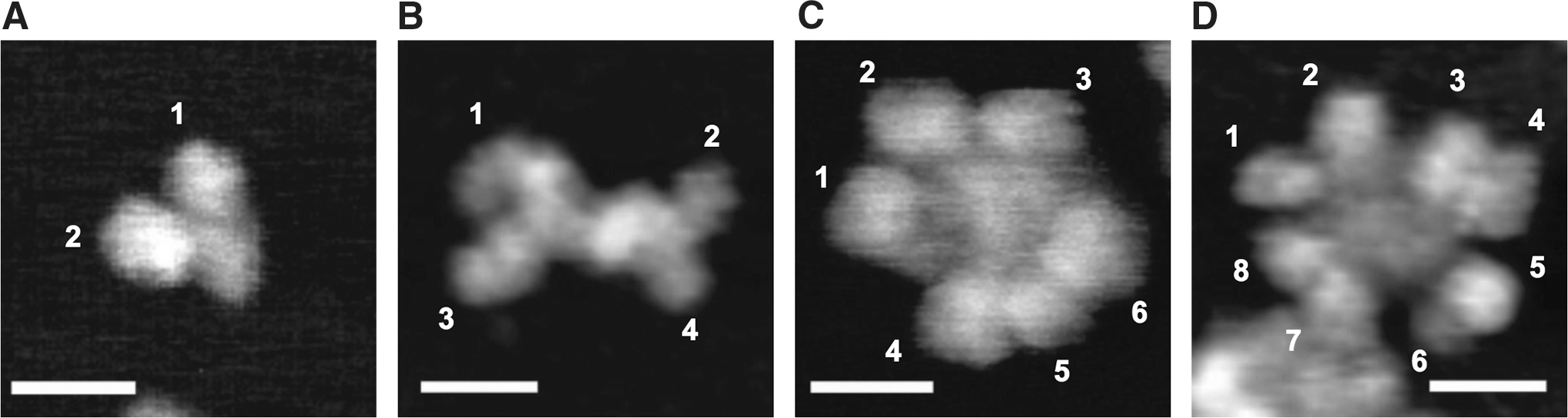
Quaternary structures of S-IgA antibodies visualized by high-speed atomic force microscopy. S-IgA antibodies were purified from nasal wash samples and separated by gel filtration chromatography. Quaternary structures of S-IgA antibodies were analyzed by high-speed atomic force microscopy.
Vaccine delivery systems targeting antigens to immune cells are also effective for modifying immune responses elicited by vaccines (41,86,87,129,136,139,140). In fact, it was recently reported that premixing vaccine antigen with a cationic hydrogel before intranasal administration enhances antigen-specific antibody responses (41,87,136,140). This cationic gel increases the viscosity of the vaccine, resulting in adhesion of vaccine to the mucosal surface in the upper respiratory tract and the prolonged retention of the vaccine in the nasal cavity. Carboxy vinyl polymer (CVP), which is a nasal mucoadhesive excipient that increases the viscosity of medicines and has been authorized as a nasal spray excipient in Japan, could be used to increase the viscosity of vaccine antigen in the nasal cavity. While the dynamics of the vaccine in the presence of CVP in the nasal cavity has not been elucidated, the addition of CVP to vaccine does enhance antibody responses in mice given an intranasal IIV (93).
Therefore, we evaluated the retention time of vaccine in the presence of CVP in the nasal cavity of macaques (109), whose nasal cavity structure is similar to that of humans (48,51,138). Rhesus macaques were sprayed intranasally with a radiolabeled WIVV derived from a A(H1N1) virus with or without CVP and then scanned with a positron emission tomography scanner for 6 h (109). In both treatments, the radioactive substance was retained in the nasal cavity through the end of the 6 h scan. However, the monkeys administered the CVP preparation that retained significantly more radioactivity in the nasal cavity (55.6% ± 2.8%) than those administered the phosphate buffered saline (PBS) preparation (32.3% ± 9.6%; p < 0.05) (Fig. 2). Intranasal administration of a WIVV with CVP induced vaccine-specific serum IgG and IgA antibodies in the cynomolgus monkey model (109). In comparison with antibody responses induced by intranasal vaccination without CVP, it was revealed that the improvement of vaccine retention in the nasal cavity in the presence of CVP enhanced the induction of IgA antibodies, not IgG, in serum. Since the level of serum IgA antibodies correlates with that of nasal IgA antibodies (3), these results suggest that the addition of CVP in the intranasal vaccination with WIVV might specifically enhance the induction of IgA antibodies in the nasal mucus.

Enhanced retention of vaccine antigen in the nasal cavity of rhesus macaques.
Thus, at least among adults with some immunological memory induced by previous infection or vaccination, intranasal administration of a WIVV without any mucosal adjuvant could successfully induce not only systemic IgG antibodies but also mucosal neutralizing polymerized S-IgA antibodies that protect against both homologous and heterologous virus. Moreover, the efficacy of a nonadjuvanted intranasal vaccination with a WIVV might be increased by the addition of a nasal mucoadhesive excipient that has already been authorized for use in humans.
Perspective
Current influenza vaccines, which are mainly IIVs composed of ether-disrupted split vaccines, effectively reduce the morbidity and mortality caused by influenza virus. In contrast, naturally induced mucosal immunity provided by S-IgA antibodies on the surface of the upper respiratory mucosa is highly effective at preventing viruses from infecting mucosal epithelial cells. IIV is inferior to natural infection in the induction of mucosal immune responses. Although LAIVs are promising candidates that elicit mucosal antibody responses, there are issues with the use of live virus. One is the exclusion of high-risk populations from the application of LAIV because of safety concerns, and another is the insufficient replication of A(H1N1) vaccine strains in human epithelial cells in upper respiratory tissue.
Intranasal vaccination with a WIVV without adjuvant could be an alternative candidate that induces both local S-IgA and systemic IgG antibodies especially in healthy adults with prior exposure to natural infection or vaccination. In our study, we used a monovalent WIVV; future studies should evaluate trivalent or quadrivalent WIVV formulations. Antigenic changes between circulating viruses and vaccine strains of influenza A(H3N2) and B viruses are currently suspected, which could undermine vaccine efficacy. The cross-neutralizing polymerized S-IgA antibodies induced by intranasal vaccination with WIVVs might mitigate this concern by providing protection at the initial site of infection.
The 2009 pandemic once again highlighted the difficulty in predicting which subtype and which strain of influenza virus will cause a new pandemic. The 2009 pandemic reminded us that an influenza virus with the ability to spread from human to human can easily and rapidly spread worldwide. At present, fatal cases in humans caused by the highly pathogenic avian influenza A(H5N1) virus (Egypt) or the avian influenza A(H7N9) virus (China) have been reported. It might be difficult to predict which of these viruses, or any other subtyped virus, will cause the next pandemic. Intranasal vaccination with representative strains could induce cross-neutralizing antibodies effective against the virus strain that circulates during the next pandemic. However, the addition of a mucosal adjuvant may be required to enhance the efficacy of an intranasal WIVV, since almost all humans are naive to these viruses. Nevertheless, intranasal administration of a WIVV without a mucosal adjuvant could successfully induce local IgA antibodies, as well as systemic IgG antibodies, and protect against seasonal influenza viruses, especially in healthy adults with immunological memory acquired from exposure to natural infection or vaccination.
Footnotes
Acknowledgments
Our works introduced in this review were supported by the grants of the Research Program on Emerging and Re-emerging Infectious Diseases from the Japanese Ministry of Health, Labor and Welfare (MHLW) and the grants of Emerging/Re-emerging Infectious Diseases Project of Japan from the Japan Agency for Medical Research and Development (AMED).
Author Disclosure Statement
No competing financial interests exist.